
Extracting HMW DNA for P. tuahiniensis Genome with Qiagen Genomic Tip Protocol
High Molecular Weight DNA for Pocillopora tuahiniensis genome
Overview
This post details my attempt at the HMW DNA extraction for the Pocillopora tuahiniensis! Zoe and I have been troubleshooting this extraction (see her posts here and here). The detailed protocol is here and here are example steps and results for P. meandrina and P. acuta.
Important notes specific to this extraction:
- We used NEB RNAse A, which is at a concentration of 1/5th the Qiagen one, so we added 95 uL instead of 19 ul to the digestion buffer
- We used Zymo Proteinase K
- XXXXXXX - amount of coral used in g
Materials and equipment
- QIAGEN Genomic-tip 100/G
- QIAGEN Genomic DNA Buffer Set
- QIAGEN RNase A (100mg/mL concentration)
- We used NEB RNAse A, which is at a concentration of 1/5th the Qiagen one, so we added 95 uL instead of 19 ul to the digestion buffer.
- QIAGEN Proteinase K
- We used Zymo Proteinase K
- DNA lo-bind tubes
- Ceramic mortar and pestle
- Forceps and scrapper (and maybe clipper, depends on your sample)
- Temperature controlled centrifuge (must go to 4 degrees C)
- Thermomixer (not 100 necessary if you have the incubator)
- TE buffer
- Rocking incubator (Incubator Genie)
- Orbital shaker
- Liquid Nitrogen
- Balance/scale
- Pipettes
- Filter pipette tips
- Wide bore pipette tips
- 100% isopropanol
- 70% ethanol
Important Notes about HMW DNA
- HMW DNA is very fragile, pipetting or vortexing can sheer it easily
- Do not vortex after extraction, to mix the samples flick gently then spin down Whenever you are pipetting the actual sample (ie, not buffers) you should use wide bore pipette tips, these are next to the kit on the Putnam shelf. They have a larger opening to the tip and are gentler
- Do not freeze the DNA, freeze thaws can break up the long fragments, keep HMW DNA in the 4 degree fridge for storage
- HMW DNA can really clump, especially in this extraction method that pellets it in a precipitation. For quantifying with the Qubit, take a top and bottom of the tube 1ul: do 2 Qubits for each sample and average them together for the whole tube concentration
Protocol
This protocol is directly copied from Zoe and Maggie. I wrote it out again in my own notebook.
Set up
- Set incubator genie to 50°C
- In a 50 mL conical, add 9.5mL of Buffer G2 (in Qiagen genomic buffer set) and 19 uL of RNAse A (95 uL of NEB RNase A at 20 mg/ml). Vortex and spin down to mix
- Clean the forceps and scrapper (and clipper if needed) with 10% bleach, DI, and 70% ethanol, then place on dry ice or in the -80 to cool down
- Put the mortar and pestle on dry ice or in the -80 to cool down
- Use the Prada or Puritz lab Thermoflask and fill with Liquid Nitrogen (you don’t need a lot, less than 1/4 full)
Tissue grinding and incubation
- Place the chilled mortar and pestle on the balance and tare
- Clip or use forceps to put the tissue piece in the pestle while on the scale, a weight between 0.5 and 1.5g (depending on how much tissue you see and how much skeleton) is good
- Take the pestle off the balance and pour in some LN2
- Wait for the liquid to boil off
- Grind the tissue until powdery, this can be hard with the skeleton. Use latex palm gloves to hold the very cold mortar and pestle. It should look something like this (example from ZD):

- Use the scrapper to scrape the tissue powder into the 50mL conical with the buffer G2 mix
- Vortex the 50mL conical
- Add 500ul of Protienase K to the conical
- Vortex the conical again
- Place the conical (on a rack) in the incubator genie at 50 degrees C and set to rocking speed 15. Incubate for 2 hours
Genomic tip extraction
- With 10 minutes left in the incubation, set the cold centrifuge to 4 degrees C and run for 10 minutes (brings temp down)
- After incubation, transfer 1mL of lysed tissue liquid into each of 8 1.5mL DNA lo-bind tubes with wide bore pipette tips (Note that the amount of tubes depends on tissue:debris ratio)
- Centrifuge at 4 degrees C for 10 minutes at 5000 rcf to pellet any extra debris
- While that is centrifuging, set up one Qiagen Genomic tip column (long column with resin filter) inside the blue holder (holds it in the tube rack) over a 50mL conical
- Add 4mL of equilibration buffer (QBT) to the resin column with filter tips and let it drip through the column, this should be done about when the centrifugation is done
- After the centrifugation there may be a pellet of debris in each tube
- Then add the supernatant from the 8 sample tubes to the resin tip with wide bore pipette tips and let drip through the column. This can take anywhere from 7 to 40 minutes depending on the sample/species etc. No pressure is applied to the column, it just drips
- When it is finished, switch out the 50mL conical underneath for a new one. Pour the liquid into a labelled waste container and dispose of the conical.
- Add 7.5mL of wash buffer (QC) and let drip through the column. This can take 5 to 15 minutes
- Pipette 5mL of elution buffer (QF) to a 5mL tube and put in the incubator genie to warm up to 50°C
- After the first wash, add another 7.5mL of wash buffer (QC) and let drip through the column
- When it’s done, transfer the resin tip to a different 50mL conical
- Add the 5mL of warmed buffer QF and let drip through
Isopropanol Precipitation of DNA
- Set up 6 DNA lo-bind tubes with all of the final labeling on them. Depending on how much liquid came out of the column you may need 6 or 7 tubes.
- Add 833ul of the eluted liquid (from the last drip out of the resin column) to each new lo-bind tube. There might not be enough for the last tube to have 833ul, so measure however much liquid you do put in that tube.
- One tube at a time, add 0.7 volumes of 100% isopropanol to each DNA low-bind tube and gently invert to mix ~10 times. For tubes that had 833ul of sample, 0.7 volumes is 583ul of isopropanol. If you have a tube with less than 833ul, calculate out what 0.7 times that volume is
- You may see DNA precipitate here, but sometimes not
- Centrifuge the lo-bind tubes for 30 minutes at 4 degrees C, 10,000 rcf
- Put a conical of 70% ethanol (you only need 1 mL per tube) in the -20 freezer to cool down while the samples are centrifuging. When ready to use, put it on an ice bucket at the bench
- Look for pellets after centrifuging, should be whitish, could be very small
- Keep the tubes in the cold centrifuge when not working with them
- One tube at a time, take them out of the centrifuge and remove most of the supernatant, not disturbing the pellet. Supernatant can go in a waste basin
- To one tube at a time, add 1mL of cold 70% ethanol and place back in the centrifuge
- Centrifuge tubes for 10 minutes at 4 degrees C 10,000 rcf
- After centrifugation, remove all of supernatant when finished, used a p20 to get small volumes out. Do the best you can to not disturb the pellet. If you cannot see a pellet, act like there is one there
- Leave the caps open and let any extra ethanol air dry for 7 minutes
- Add 50ul of TE buffer to each tube, gently
- Place each tube in the Thermomixer for 1 hour at 55 degrees C no shaking
- Place the tubes in a tube rack on the orbital shaker for overnight, 200rpm. Qubit and TapeStation the next day. If it is a Friday, put the tubes on the shaker as long as you can, then place them in the 4 degree C fridge for the weekend. Before Qubit the next Monday, put them on the shaker for an hour
QC
What is our goal for amount of DNA?
-
From the sequencing facility (BYU, through genohub): “Three micrograms of DNA should be provided to enable library construction and Blue-Pippin size selection for large fragment whole genome libraries”
-
(Danielle’s post said: For PacBio we need 10-20ug of genomic DNA (as quantified by Qubit) in less than 400ul. Quality of the DNA should be over 40kb in size, though we can remove smaller fragments.)
Qubit
- Use the broad range DNA assay and protocol
- Quantify both the top and bottom of each tube and then use the averages of each tube for the final concentration
TapeStation
- For accurate sizing, we use the TapeStation instead of a gel. However it still isn’t the absolute best, the best thing for HMW DNA is a pulse-phase gel that’s run super slowly and alternates the position of the current to separate out the large fragments. You can look at pictures, pretty cool. However, we don’t have the type of gel box for that!
- Use the Genomic DNA screentapes and reagent/ladder. This reads from 48,000bp to 100bp
- Follow the protocol
- Your goal is to have a huge peak at the 60,000bp size or around there, and very minimal smearing elsewhere
- Good trace:
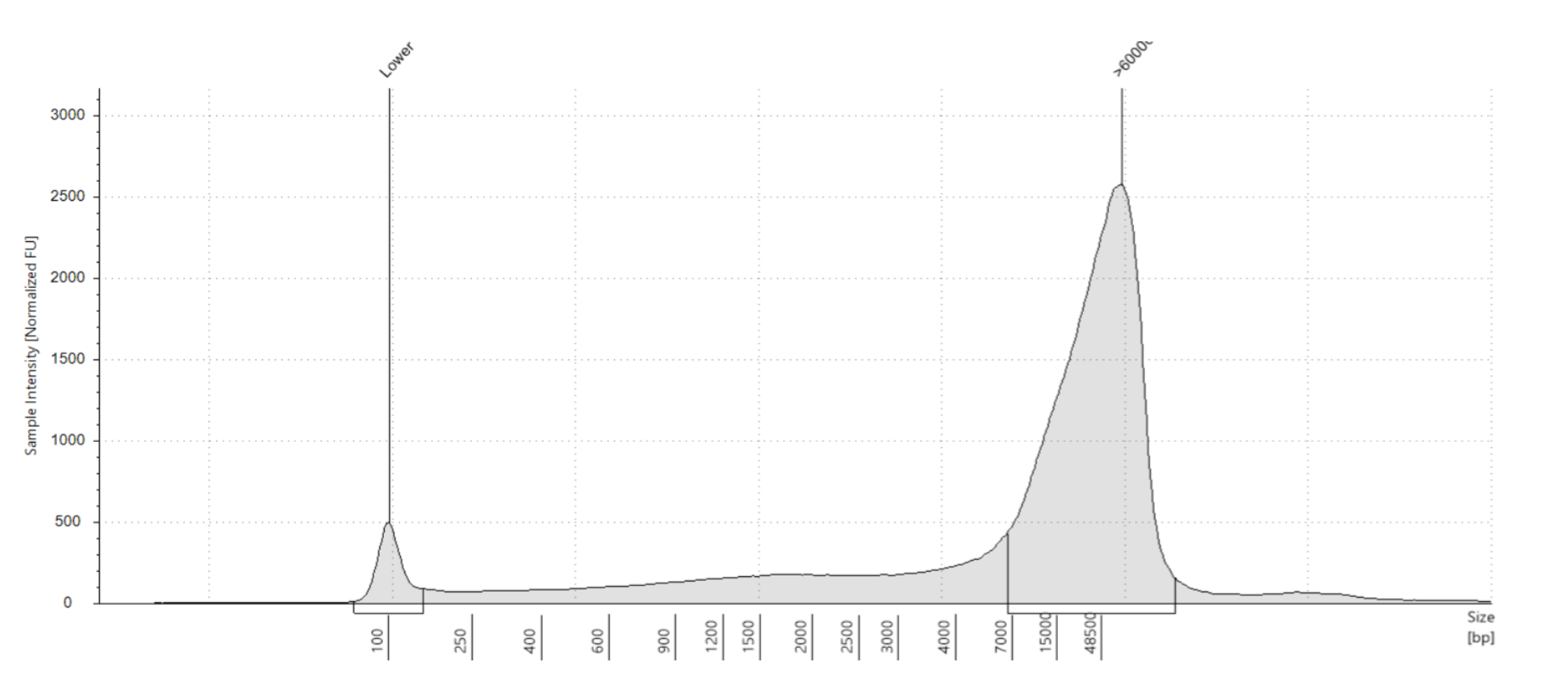
- Bad trace:
