
Point Judith Oyster Nutrient DNA methylation
Point Judith oyster DNA methylation
As a lab, we have been working on an oyster paper that details the responses of Eastern oysters to differing nutrient regimes. Here is the project github page.
ES ran the methylation data through the nf-core methylseq pipeline, but there are some discrepancies in the QC outputs that we need to resolve. Methylseq doesn’t appear to be recognizing the adapter bp and so isn’t properly trimming them. Here is ES notebook post detailing her methylseq runs. To address this, I will run the methylseq pipeline but run each step individually (as opposed to the steps being run in the pipeline wrapper).
Set up data in my directory
Original data lives here: /data/putnamlab/KITT/hputnam/20200119_Oyst_Nut/MBDBS
Make new folder in my own directory
cd /data/putnamlab/jillashey
mkdir Oys_Nutrient
cd Oys_Nutrient
mkdir MBDBS RNASeq
cd MBDBS
mkdir data scripts fastqc_results
cd data
mkdir raw trim
cd ../fastqc_results
mkdir raw trim
Copy data over into my directory
There are 24 total files, but 12 total samples (each sample has a R1 and R2 file).
cd /data/putnamlab/KITT/hputnam/20200119_Oyst_Nut/MBDBS
cp *fastq.gz /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
Count number of reads per file
Number of reads can be counted by counting the number of ‘@__’ terms there are in the file, as each one corresponds to one read.
zgrep -c "@M00" *fastq.gz
HPB10_S44_L001_R1_001.fastq.gz:1036453
HPB10_S44_L001_R2_001.fastq.gz:1036453
HPB11_S45_L001_R1_001.fastq.gz:976844
HPB11_S45_L001_R2_001.fastq.gz:976844
HPB12_S46_L001_R1_001.fastq.gz:1541665
HPB12_S46_L001_R2_001.fastq.gz:1541665
HPB1_S35_L001_R1_001.fastq.gz:965843
HPB1_S35_L001_R2_001.fastq.gz:965843
HPB2_S36_L001_R1_001.fastq.gz:990538
HPB2_S36_L001_R2_001.fastq.gz:990538
HPB3_S37_L001_R1_001.fastq.gz:953246
HPB3_S37_L001_R2_001.fastq.gz:953246
HPB4_S38_L001_R1_001.fastq.gz:864752
HPB4_S38_L001_R2_001.fastq.gz:864752
HPB5_S39_L001_R1_001.fastq.gz:923141
HPB5_S39_L001_R2_001.fastq.gz:923141
HPB6_S40_L001_R1_001.fastq.gz:909119
HPB6_S40_L001_R2_001.fastq.gz:909119
HPB7_S41_L001_R1_001.fastq.gz:1053144
HPB7_S41_L001_R2_001.fastq.gz:1053144
HPB8_S42_L001_R1_001.fastq.gz:923398
HPB8_S42_L001_R2_001.fastq.gz:923398
HPB9_S43_L001_R1_001.fastq.gz:1067829
HPB9_S43_L001_R2_001.fastq.gz:1067829
Run fastqc on raw data
In scripts folder: nano fastqc_raw.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_raw_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_raw_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/raw
done
multiqc --interactive fastqc_results
sbatch fastqc_raw.sh; Submitted batch job 239141
Copy multiQC report to Cvir github and look at results
On local computer:
scp jillashey@ssh3.hac.uri.edu:/data/putnamlab/jillashey/Oys_Nutrient/MBDBS/multiqc_report.html /Users/jillashey/Desktop/PutnamLab/Repositories/Cvir_Nut_Int/output/MBDBS/JA
- This is ES initial report: https://github.com/hputnam/Cvir_Nut_Int/blob/master/output/MBDBS/initial_multiqc_report.html
- This is my initial report: https://github.com/hputnam/Cvir_Nut_Int/blob/master/output/MBDBS/JA/raw_multiqc_report.html
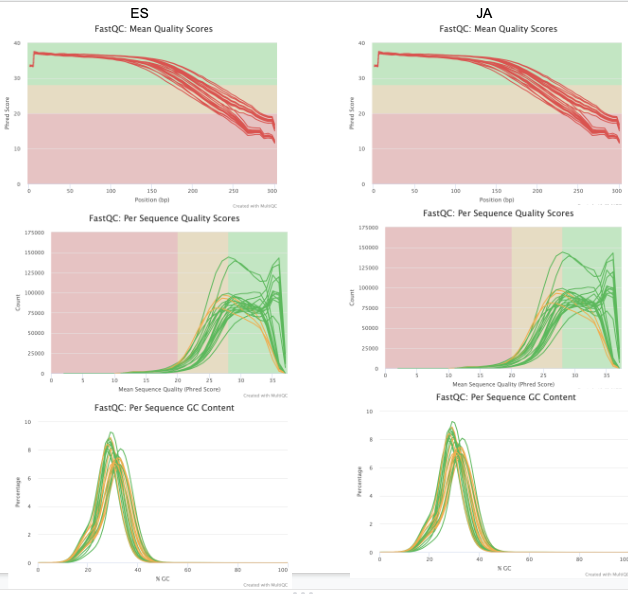
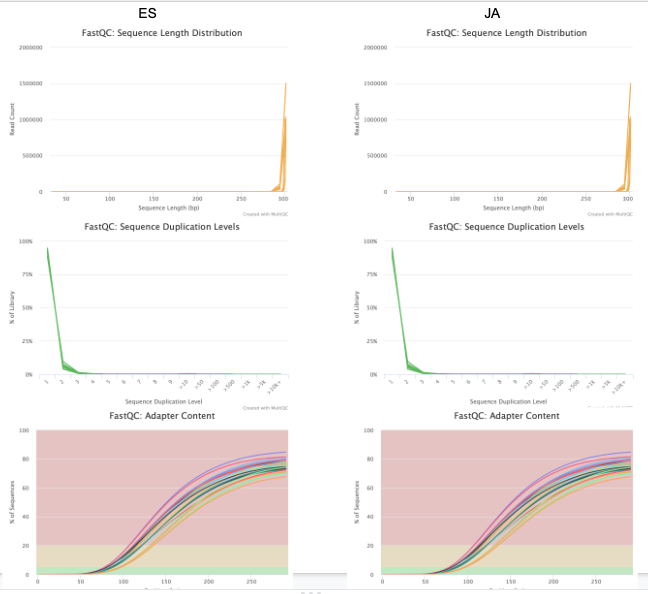
Mine & ES report seem to be the same. That’s a good sign (I believe)! Now can move onto trimming w/ Trim Galore.
Trim sequences
Going to trim w/ Trim Galore, as this is what methylseq uses for trimming purposes.
Two versions of trim galore on URI Andromeda: all/Trim_Galore/0.6.6-GCC-10.2.0-Python-2.7.18 and all/Trim_Galore/0.6.7-GCCcore-11.2.0. I’m going to use the newest one.
I’m going to do several iterations of trimming so I’m going to make different trimming folders in my data and fastqc_results folder.
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results
mkdir trim1 trim2 trim3 trim4 trim5
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data
mkdir trim1 trim2 trim3 trim4 trim5
Attempt 1
I’m not going to add any sort of cutoffs for this initial trimming run.
In scripts folder: nano trim_galore1.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore1_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore1_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
module load Trim_Galore/0.6.7-GCCcore-11.2.0
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim1/
done
sbatch trim_galore1.sh; Submitted batch job 240029
For each read, the script generates a new/trimmed .fq.gz file and a trimming report .txt file. Here’s an example of what the .txt file looks like for sample HPB10_S44_L001_R1_001:
Using Illumina adapter for trimming (count: 804887). Second best hit was Nextera (count: 4)
Adapter sequence: 'AGATCGGAAGAGC' (Illumina TruSeq, Sanger iPCR; auto-detected)
Maximum trimming error rate: 0.1 (default)
Minimum required adapter overlap (stringency): 1 bp
Minimum required sequence length for both reads before a sequence pair gets removed: 20 bp
Output file will be GZIP compressed
This is cutadapt 3.5 with Python 3.9.6
Command line parameters: -j 1 -e 0.1 -q 20 -O 1 -a AGATCGGAAGAGC HPB10_S44_L001_R1_001.fastq.gz
Processing reads on 1 core in single-end mode ...
Finished in 77.59 s (75 µs/read; 0.80 M reads/minute).
=== Summary ===
Total reads processed: 1,036,453
Reads with adapters: 984,111 (94.9%)
Reads written (passing filters): 1,036,453 (100.0%)
Total basepairs processed: 311,706,539 bp
Quality-trimmed: 51,156,210 bp (16.4%)
Total written (filtered): 173,587,926 bp (55.7%)
=== Adapter 1 ===
Sequence: AGATCGGAAGAGC; Type: regular 3'; Length: 13; Trimmed: 984111 times
Minimum overlap: 1
No. of allowed errors:
1-9 bp: 0; 10-13 bp: 1
Bases preceding removed adapters:
A: 32.8%
C: 12.2%
G: 21.1%
T: 33.8%
none/other: 0.0%
Attempt 2
In ES notebook post, she ran a couple of different iterations of methylseq where she used different cutoffs for trimming.
PJ_methylseq1.sh: --clip_r1 10 \ --clip_r2 10 \ --three_prime_clip_r1 10 --three_prime_clip_r2 10 \- From ES notebook: Run this first to assess m-bias and then decide if we need more trial runs. See Emma Strand and Kevin Wong’s notebook posts for methylation scripts and how they dealt with these issues.
PJ_methylseq2.sh: --clip_r1 10 \ --clip_r2 10 \ --three_prime_clip_r1 20 --three_prime_clip_r2 20 \- From ES notebook: Because the run time seemed oddly short, I wanted to run this again to be sure it worked.
PJ_methylseq3.sh: --clip_r1 150 \ --clip_r2 150 \ --three_prime_clip_r1 150 --three_prime_clip_r2 150 \- From ES notebook: We think that methylseq isn’t properly recognizing the adapters because 1.) the multiqc report says 300 bp length when usually for methylation data we use 2x150 bp. 2.) If the program isn’t trimming the adapter fully that would explain the extreme m-bias, adapter content, and decreased quality all after 150 bp length. 3.) Usually Illumina adapters are ~150 bp of the read length and are not included in multiqc reports (if recognized appropriately). I’m cutting 150 here to see if this makes a difference but unsure if we trust methylseq if it’s not recognizing the adapters (i.e. what other problems might we have and don’t see yet?)
Parameters
--clip_R1removes XX bps from the 5’ end of read 1.--clip_R2removes XX bps from the 5’ end of read 2.--three_prime_clip_R1removes XX bp after the 3’ end of read 1 AFTER adapter/quality trimming has been performed--three_prime_clip_R2removes XX bp after the 3’ end of read 2 AFTER adapter/quality trimming has been performed
Running her first iteration (--clip_r1 10 \ --clip_r2 10 \ --three_prime_clip_r1 10 --three_prime_clip_r2 10 \). I’m clipping 10 bp from the 5’ end of reads 1 and 2. After adapter/quality trimming has been done, 10 bp will also be clipped from the 3’ end of reads 1 and 2.
In scripts folder: nano trim_galore2.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore2_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore2_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
module load Trim_Galore/0.6.7-GCCcore-11.2.0
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz --clip_r1 10 --clip_r2 10 --three_prime_clip_r1 10 --three_prime_clip_r2 10 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim2/
done
sbatch trim_galore2.sh; Submitted batch job 241069
Attempt 3
Running her second iteration (--clip_r1 10 \ --clip_r2 10 \ --three_prime_clip_r1 20 --three_prime_clip_r2 20 \)
In scripts folder: nano trim_galore3.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore3_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore3_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
module load Trim_Galore/0.6.7-GCCcore-11.2.0
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz --clip_r1 10 --clip_r2 10 --three_prime_clip_r1 20 --three_prime_clip_r2 20 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim3/
done
sbatch trim_galore3.sh; Submitted batch job 241074
Attempt 4
Running her third iteration: --clip_r1 150 \ --clip_r2 150 \ --three_prime_clip_r1 150 --three_prime_clip_r2 150 \
In scripts folder: nano trim_galore4.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore4_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore4_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
module load Trim_Galore/0.6.7-GCCcore-11.2.0
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz --clip_r1 150 --clip_r2 150 --three_prime_clip_r1 150 --three_prime_clip_r2 150 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim4/
done
sbatch trim_galore4.sh; Submitted batch job 241075
This failed - error file said that “The 5’ clipping value for read 1 should have a sensible value (> 0 and < read length)”. So the value should be greater than 0 and less than the read length (300?). But isn’t our read length 300 and 150 is less than 300? Changing clip_r1, clip_r2, three_prime_clip_r1, and three_prime_clip_r2 to 100 instead of 150 and seeing how that works. Submitted batch job 241088. Failed agian, same error as before.
In the trim galore source code, it looks like trimming is only possible if its between 0 and less than 100. See code here:
if (defined $clip_r1){ # trimming 5' bases of read 1
unless ($clip_r1 > 0 and $clip_r1 < 100){
die "The 5' clipping value for read 1 should have a sensible value (> 0 and < read length)\n\n";
}
}
if (defined $clip_r2){ # trimming 5' bases of read 2
unless ($clip_r2 > 0 and $clip_r2 < 100){
die "The 5' clipping value for read 2 should have a sensible value (> 0 and < read length)\n\n";
}
}
### Trimming at the 3' end
if (defined $three_prime_clip_r1){ # trimming 3' bases of read 1
unless ($three_prime_clip_r1 > 0 and $three_prime_clip_r1 < 100){
die "The 3' clipping value for read 1 should have a sensible value (> 0 and < read length)\n\n";
}
}
if (defined $three_prime_clip_r2){ # trimming 3' bases of read 2
unless ($three_prime_clip_r2 > 0 and $three_prime_clip_r2 < 100){
die "The 3' clipping value for read 2 should have a sensible value (> 0 and < read length)\n\n";
}
So I don’t think I can trim more than 100 bp. Let’s try 99 bp in trim_galore4.sh just to see if it will work. Submitted batch job 241090.
Attempt 5
Based on the fastqc results, it looks like the reads are still relatively low quality at the end of the reads. I’m going to add the --quality flag and set the Phred score to 30.
In scripts folder: nano trim_galore5.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore5_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore5_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
module load Trim_Galore/0.6.7-GCCcore-11.2.0
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz --quality 30 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim5/
done
sbatch trim_galore5.sh; Submitted batch job 241165
Run Fastqc on trimmed data
Attempt 1
In scripts folder: nano fastqc_trim1.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim1_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim1_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim1/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim1
done
multiqc --interactive fastqc_results/trim1
sbatch fastqc_trim1.sh; Submitted batch job 241058
Attempt 2
In scripts folder: nano fastqc_trim2.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim2_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim2_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim2/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim2
done
multiqc --interactive fastqc_results/trim2
sbatch fastqc_trim2.sh; Submitted batch job 241100
Attempt 3
In scripts folder: nano fastqc_trim3.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim3_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim3_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim3/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim3
done
multiqc --interactive fastqc_results/trim3
sbatch fastqc_trim3.sh; Submitted batch job 241125
Attempt 4
In scripts folder: nano fastqc_trim4.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim4_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim4_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim4/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim4
done
multiqc --interactive fastqc_results/trim4
sbatch fastqc_trim4.sh; Submitted batch job 241148
Attempt 5
In scripts folder: nano fastqc_trim5.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim5_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim5_output" #once your job is completed, any final job report comments will be put in this file
source /usr/share/Modules/init/sh # load the module function
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim5/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim5
done
multiqc --interactive fastqc_results/trim5
sbatch fastqc_trim5.sh; Submitted batch job 241185
Look at fastqc results
Trimming overview
As a reminder, these are the parameters from each trimming run:
| Trim1 | Trim2 | Trim3 | Trim4 | Trim5 | |
|---|---|---|---|---|---|
paired |
Yes | Yes | Yes | Yes | Yes |
clip_r1 |
NA | 10 | 10 | 99 | NA |
clip_r2 |
NA | 10 | 10 | 99 | NA |
three_prime_clip_r1 |
NA | 10 | 20 | 99 | NA |
three_prime_clip_r2 |
NA | 10 | 20 | 99 | NA |
quality |
NA | NA | NA | NA | 30 |
Fastqc plots
ES, JA trim1, JA trim2, JA trim3
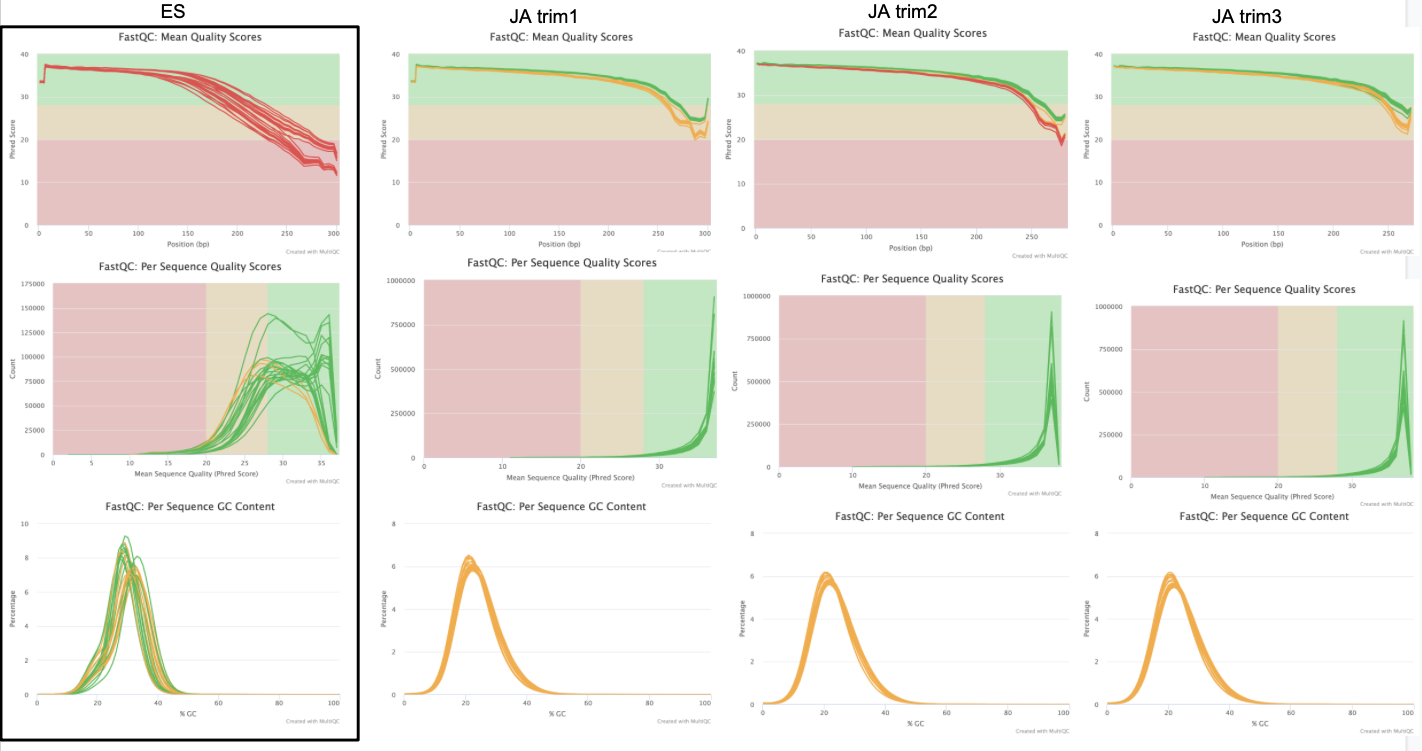
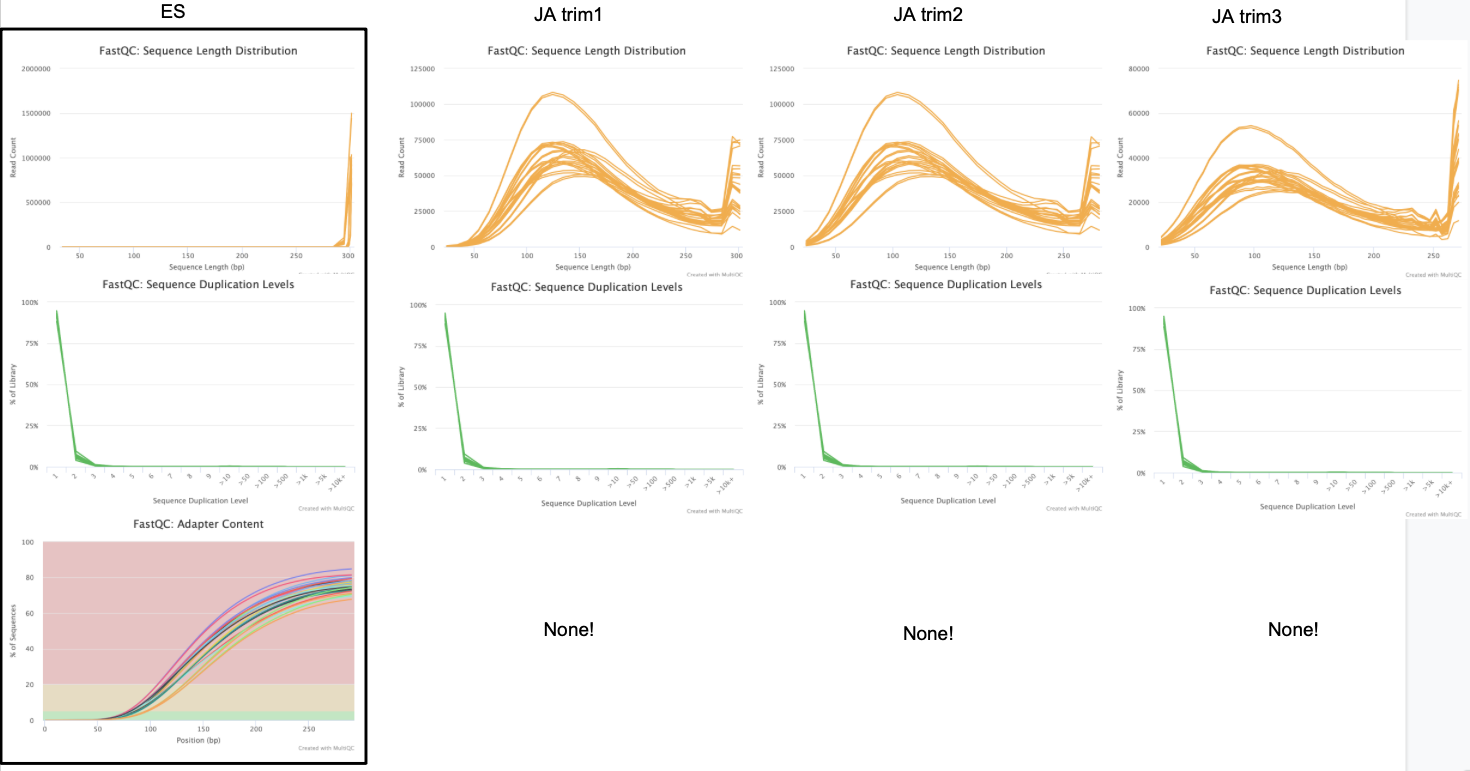
JA trim4, JA trim5
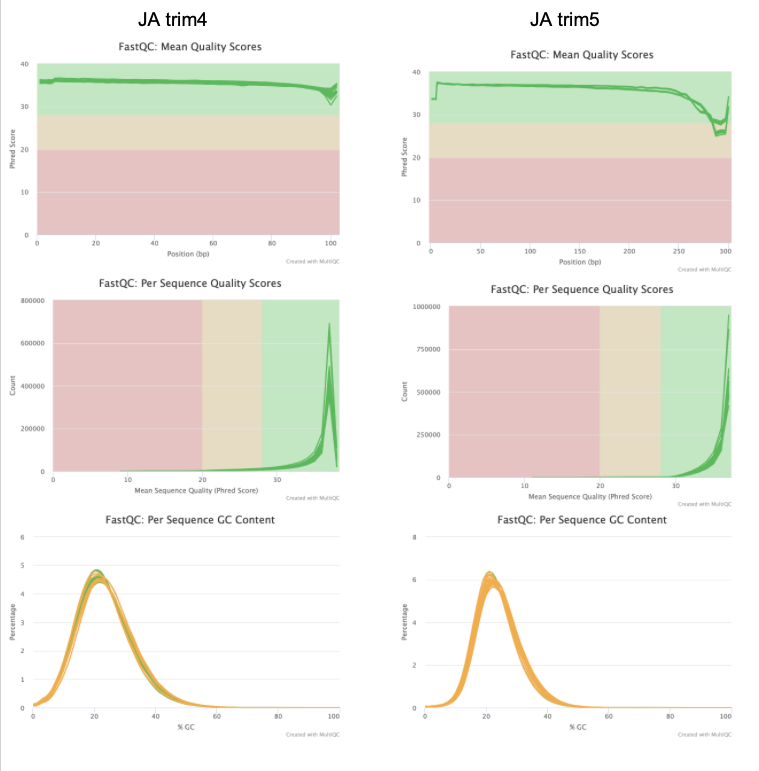
Discuss results w/ lab
Bismark
Bismark is a program to map bisulfite treated sequencing reads to a genome of interest and perform methylation calls in a single step. Methylseq uses Bismark in its workflow.
Bismark needs Bowtie2 or HISAT2 and Samtools to properly run.
Prepare genome
First step is indexing the genome to prepare it for alignment. The genome needs to be either a .fa, .fa.gz, .fasta or .fasta.gz file extension. The genome in /data/putnamlab/shared/Oyst_Nut_RNA/references/ is a .fna file, so I need to rename it. I also need to download the genome files because I can’t access them in the shared folder.
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/GCF_002022765.2_C_virginica-3.0_genomic.fna.gz
gunzip GCF_002022765.2_C_virginica-3.0_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/002/022/765/GCF_002022765.2_C_virginica-3.0/GCF_002022765.2_C_virginica-3.0_genomic.gff.gz
gunzip GCF_002022765.2_C_virginica-3.0_genomic.gff.gz
cp GCF_002022765.2_C_virginica-3.0_genomic.fna GCF_002022765.2_C_virginica-3.0_genomic.fa
In scripts folder: nano bismark_genome_prep.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_genome_prep_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_genome_prep_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load Bowtie2/2.4.4-GCC-11.2.0
bismark_genome_preparation --verbose /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs
echo "Bismark genome prep complete" $(date)
Submitted batch job 246354
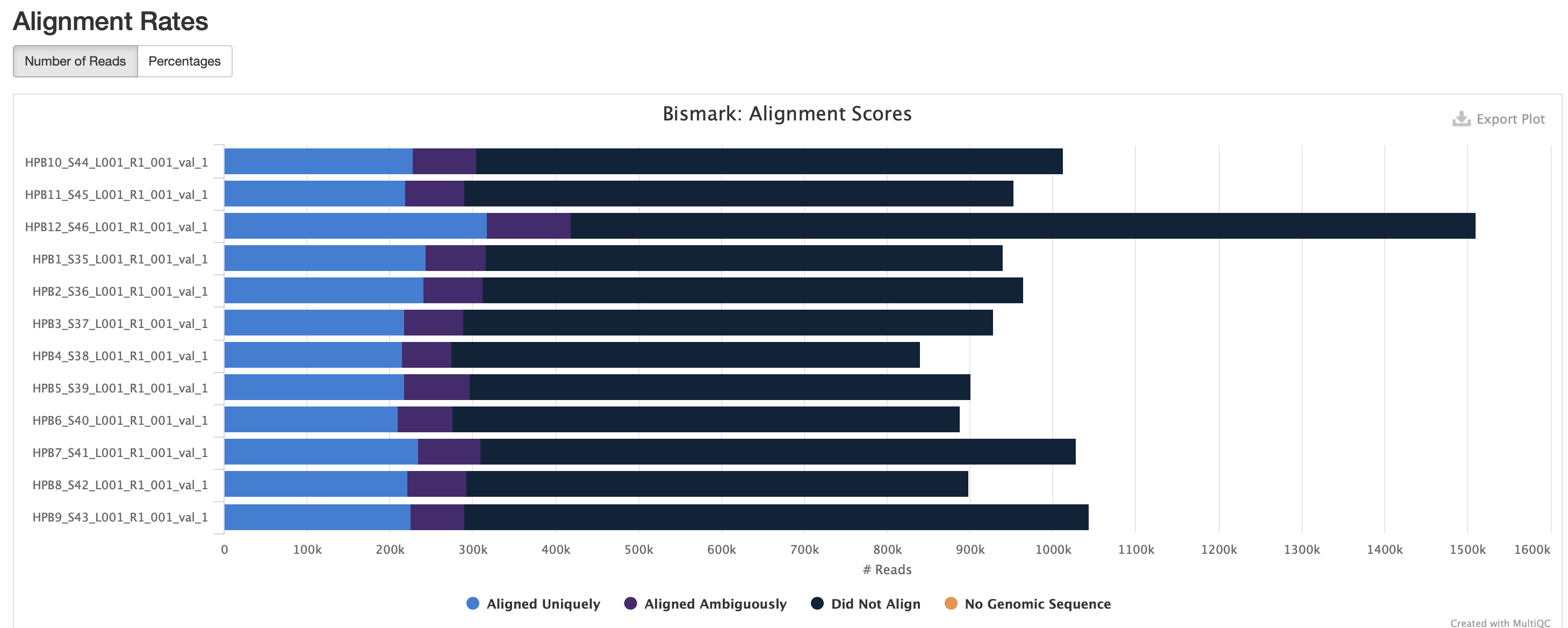
Align reads
I’m going to use the data from trim5 for now. No adapter content is present and the quality is high for all reads.
In scripts folder: nano bismark_align.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_align_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_align_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load Bowtie2/2.4.4-GCC-11.2.0
echo "Starting Bismark alignment" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim5
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
bismark --multicore 10 --bam --non_directional --output_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/alignment --temp_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/temp --unmapped --ambiguous --genome /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs -1 ${file}_L001_R1_001_val_1.fq.gz -2 ${file}_L001_R2_001_val_2.fq.gz
done
echo "Bismark alignment complete!" $(date)
Submitted batch job 246358
Should I include the score_min and/or the relax_mismatches agrument? Discuss w/ lab
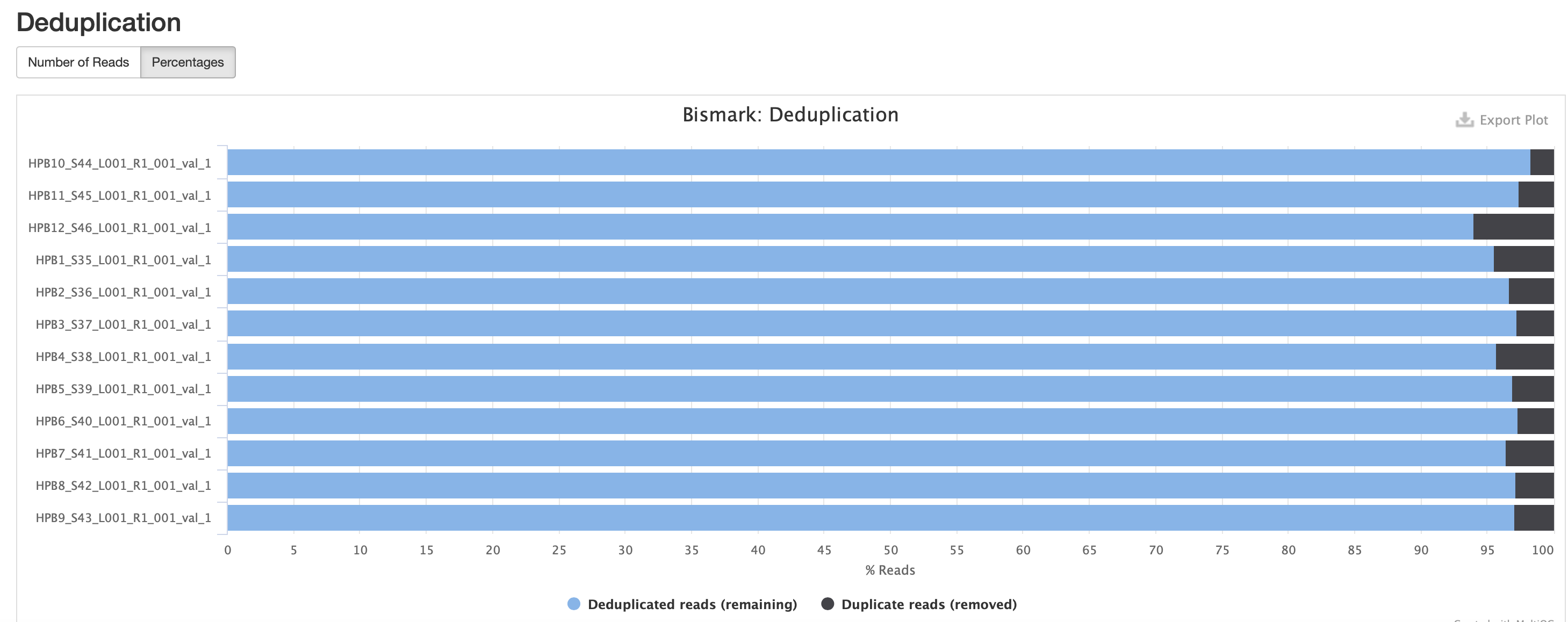
Deduplicate alignments
It looks like the paired reads were ‘merged’ into one bam file.
In scripts folder: nano bismark_deduplicate.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_deduplicate_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_deduplicate_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
echo "Starting Bismark deduplication" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/alignment
for file in *bam
do
deduplicate_bismark --paired --bam --output_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/deduplicate $file
done
echo "Bismark deduplication complete!" $(date)
Submitted batch job 246895
Do the files need to be sorted or indexed via Samtools before moving on to the next step? Discuss w/ lab
I’m going to sort and index using samtools for downstream analysis. Based on Javie’s code here.
In scripts folder: nano sort_index.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="sort_index_error" #if your job fails, the error report will be put in this file
#SBATCH --output="sort_index_output" #once your job is completed, any final job report comments will be put in this file
module load SAMtools/1.16.1-GCC-11.3.0
echo "Starting to sort and index the deduplicated bam files" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/deduplicate
for file in *deduplicated.bam
do
samtools sort $file -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/deduplicate/${file}_dedup.sort.bam
done
for file in *dedup.sort.bam
do
samtools index $file
done
echo "Sorting and indexing complete!" $(date)
Submitted batch job 246904
Extract methylation calls
Should I use the deduplicated.bam or dedup.sort.bam? I’m going to use the deduplicated.bam for now.
First, make a folder for the results
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark
mkdir methyl_extract
Now in the scripts folder: nano bismark_methyl_extract.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_methyl_extract_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_methyl_extract_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load SAMtools/1.16.1-GCC-11.3.0
echo "Starting to extract methylation calls" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/deduplicate
for file in *deduplicated.bam
do
bismark_methylation_extractor --paired-end --bedGraph --scaffolds --cytosine_report --genome_folder /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs --output /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/methyl_extract $file
done
echo "Methylation extraction complete!" $(date)
Submitted batch job 246909. This took about 2.5 hours
Organize directory
mkdir CHH CHG CpG
mv CHG_* CHG
mv CHH_* CHH
mv CpG_* CpG
Sample report
There are two reports to generate - bismark2report and bismark2summary. bismark2report generates a seperate HTML file for each sample, but bismark2summary reports a summary of all the samples. Both take different arguments.
bismark2report
This module requires an alignment report file and has optional arguments for deduplication report, splitting report, m-bias report and nucleotide report. The alignment report files were generated in the Bismark alignment step and the dedup, splitting and m-bias reports were generated in the methylation extraction step. I’m going to make a new directory for the report files.
I’m going to move the alignment report files into the methyl_extract folder to make it easier to run the code.
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark
mkdir logs
# alignment reports
cd alignment
mv *bismark_bt2_PE_report.txt ../logs
# deduplication reports
cd ../deduplicate/
mv *val_1_bismark_bt2_pe.deduplication_report.txt ../logs
# splitting & mbias reports
cd ../methyl_extract/
mv *deduplicated.M-bias.txt ../logs
mv *deduplicated_splitting_report.txt ../logs
Now run the report. I’m going to run each one individually because it won’t take that much time.
module load Bismark/0.23.1-foss-2021b
bismark2report --alignment_report HPB10_S44_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB10_S44_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB10_S44_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB10_S44_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB11_S45_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB11_S45_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB11_S45_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB11_S45_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB12_S46_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB12_S46_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB12_S46_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB12_S46_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB1_S35_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB1_S35_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB1_S35_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB1_S35_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB2_S36_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB2_S36_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB2_S36_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB2_S36_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB3_S37_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB3_S37_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB3_S37_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB3_S37_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB4_S38_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB4_S38_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB4_S38_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB4_S38_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB5_S39_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB5_S39_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB5_S39_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB5_S39_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB6_S40_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB6_S40_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB6_S40_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB6_S40_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB7_S41_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB7_S41_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB7_S41_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB7_S41_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB8_S42_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB8_S42_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB8_S42_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB8_S42_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2report --alignment_report HPB9_S43_L001_R1_001_val_1_bismark_bt2_PE_report.txt --dedup_report HPB9_S43_L001_R1_001_val_1_bismark_bt2_pe.deduplication_report.txt --splitting_report HPB9_S43_L001_R1_001_val_1_bismark_bt2_pe.deduplicated_splitting_report.txt --mbias_report HPB9_S43_L001_R1_001_val_1_bismark_bt2_pe.deduplicated.M-bias.txt
bismark2summary
Which bam files should I use (ie ones from the alignment directory or the deduplicate directory)? I’m going to use the ones in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/alignment. It needs the associated alignment report files and the bam files. I’m going to copy the bam files into the logs file.
module load Bismark/0.23.1-foss-2021b
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/alignment
cp *bam ../logs/
cd ../logs
bismark2summary *bam
MultiQC
In the logs directory, run MultiQC on the data
module load MultiQC/1.9-intel-2020a-Python-3.8.2
multiqc -f --filename multiqc_report . \
-m custom_content -m picard -m qualimap -m bismark -m samtools -m preseq -m cutadapt -m fastqc
Secure copy the file to the github repo
MultiQC results
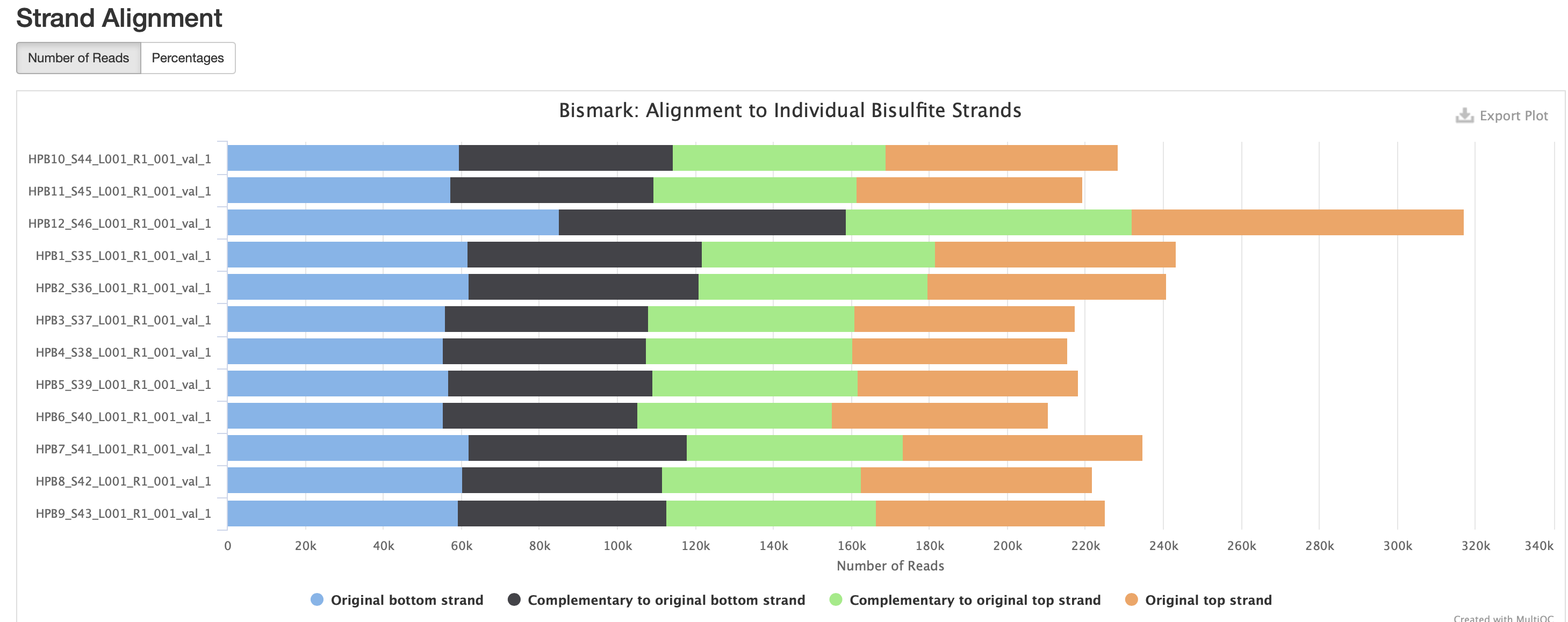
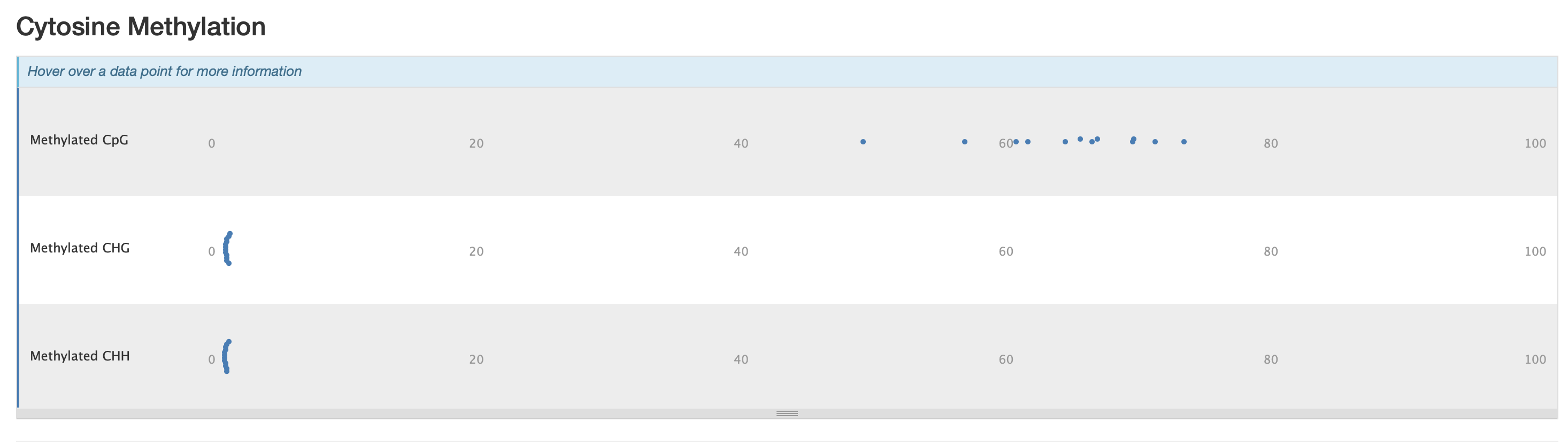
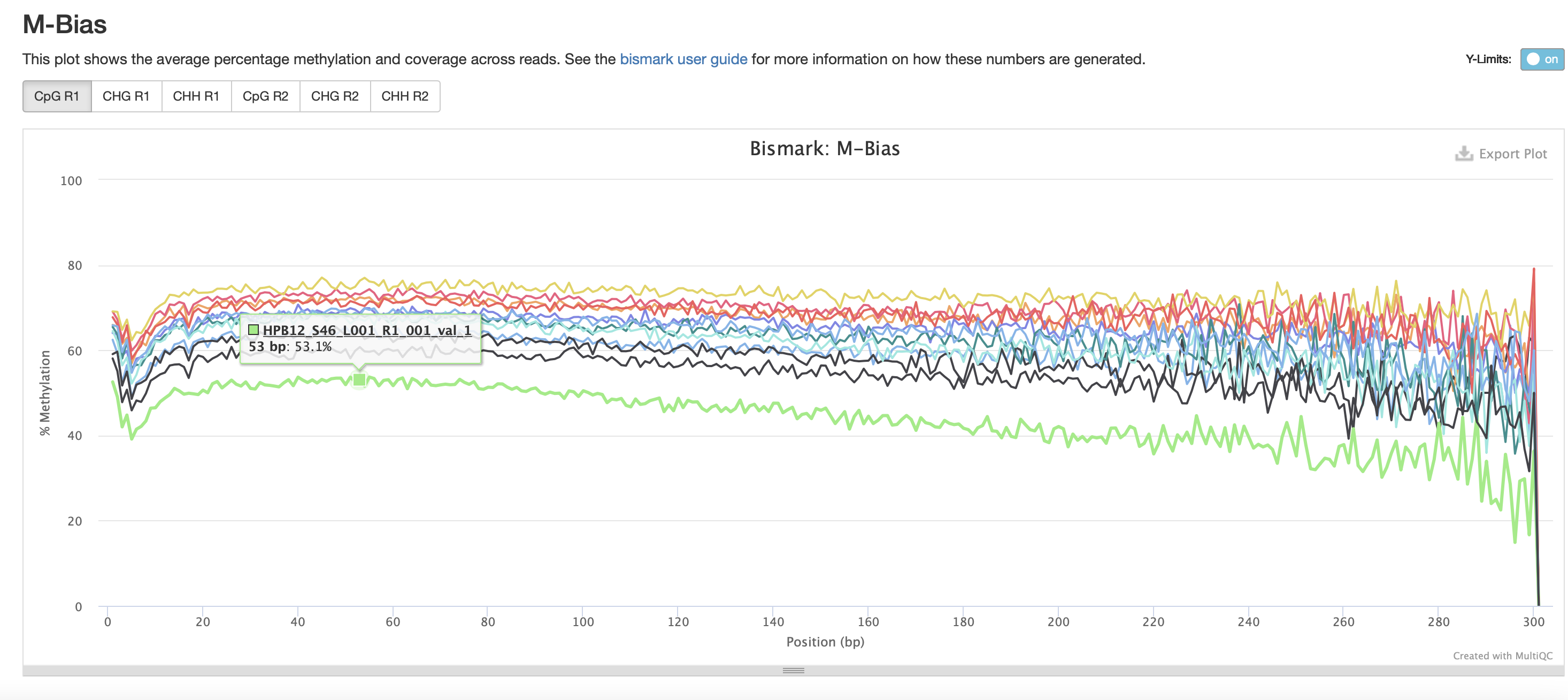
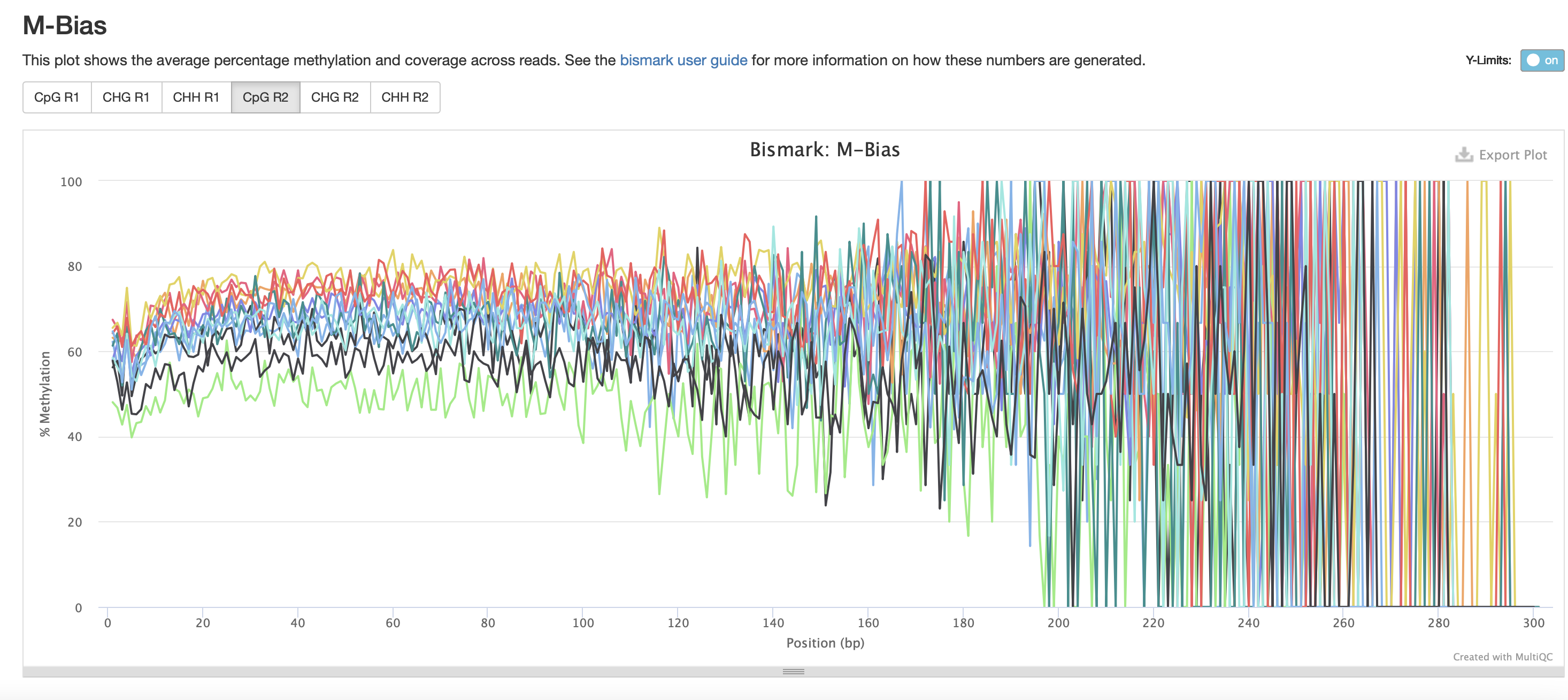
The M-bias gets pretty wacky towards the end of the read, similar to one of the methyseq results from ES.
STILL NEED TO DO QUALIMAP FOR ALIGNMENT QC AND PRESEQ FOR SAMPLE COMPLEXITY (as detailed in the methylseq pipline)!!!
Thoughts
- Arguments that I should’ve used?
--relax_mismatches- seems to only be in the methylseq pipeline--score_min- Sets a function governing the minimum alignment score needed for an alignment to be considered “valid” (i.e. good enough to report). This is a function of read length. For instance, specifying L,0,-0.2 sets the minimum-score function f to f(x) = 0 + -0.2 * x, where x is the read length.- Javie has code here where he evaluated
--score_minat -0.2, -0.4, -0.6, -0.9, -1.2, and -1.5. He ended up going with the-score_min L,0,-0.9 - Include in alignment step
- Javie has code here where he evaluated
20230416
After talking with the lab about the results, we decided to do another trimming iteration. Looking at the sequence quality histogram from the raw QC file, it seems that the quality drips below a 20 phred score around 227 bp. I’m going to trim the 5’ end by 75 bp and then proceed w/ Bismark.
Trim sequences
Make folders from trim6 iteration
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results
mkdir trim6
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data
mkdir trim6
Attempt 6
In scripts folder: nano trim_galore6.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore6_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore6_output" #once your job is completed, any final job report comments will be put in this file
module load Trim_Galore/0.6.7-GCCcore-11.2.0
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz --clip_r1 75 --clip_r2 75 --quality 30 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim6/
done
Submitted batch job 251641
Run Fastqc on trimmed data
Attempt 6
In scripts folder: nano fastqc_trim6.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim6_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim6_output" #once your job is completed, any final job report comments will be put in this file
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim6/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim6
done
multiqc --interactive fastqc_results/trim6
Submitted batch job 251642
FastQC results for trim6 iteration
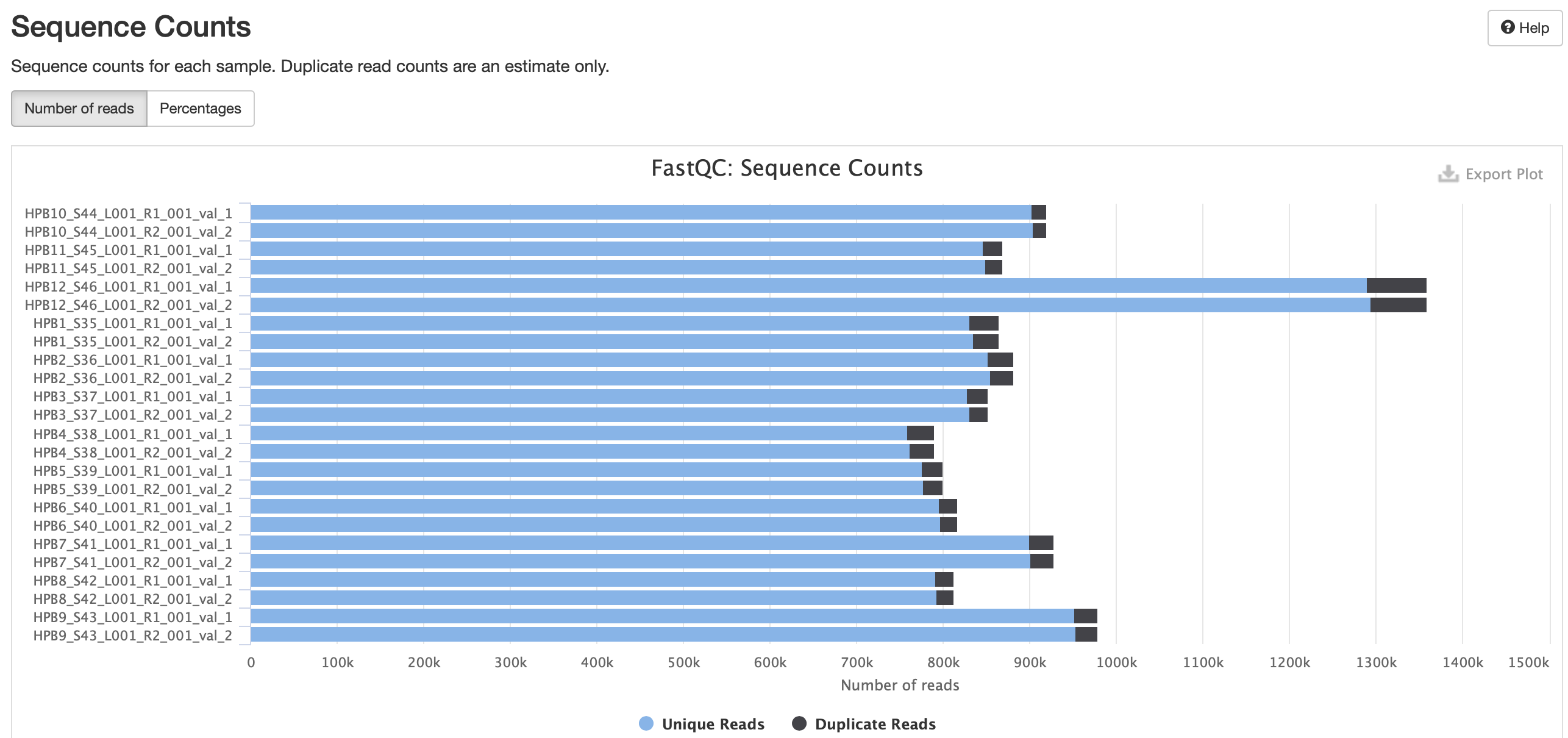
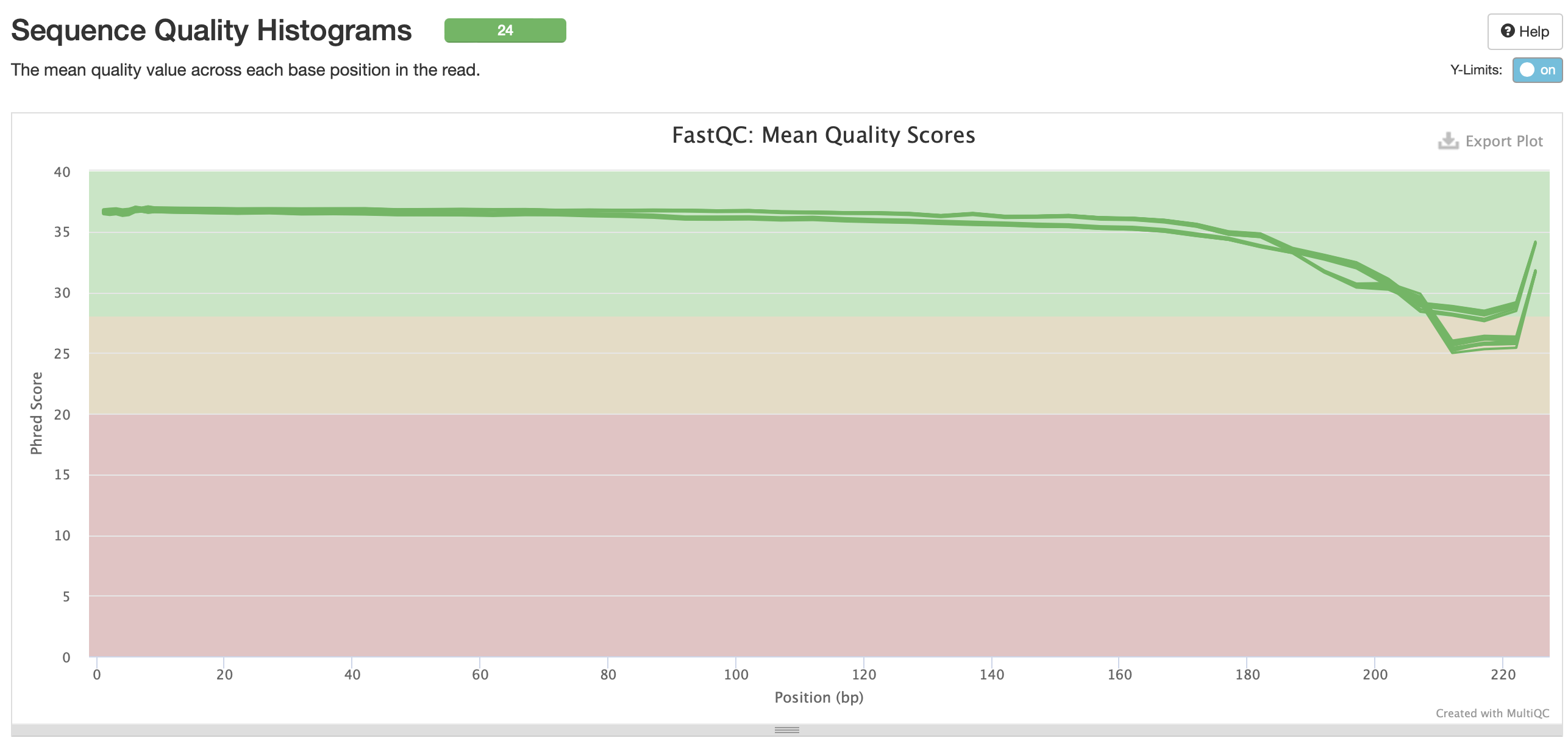
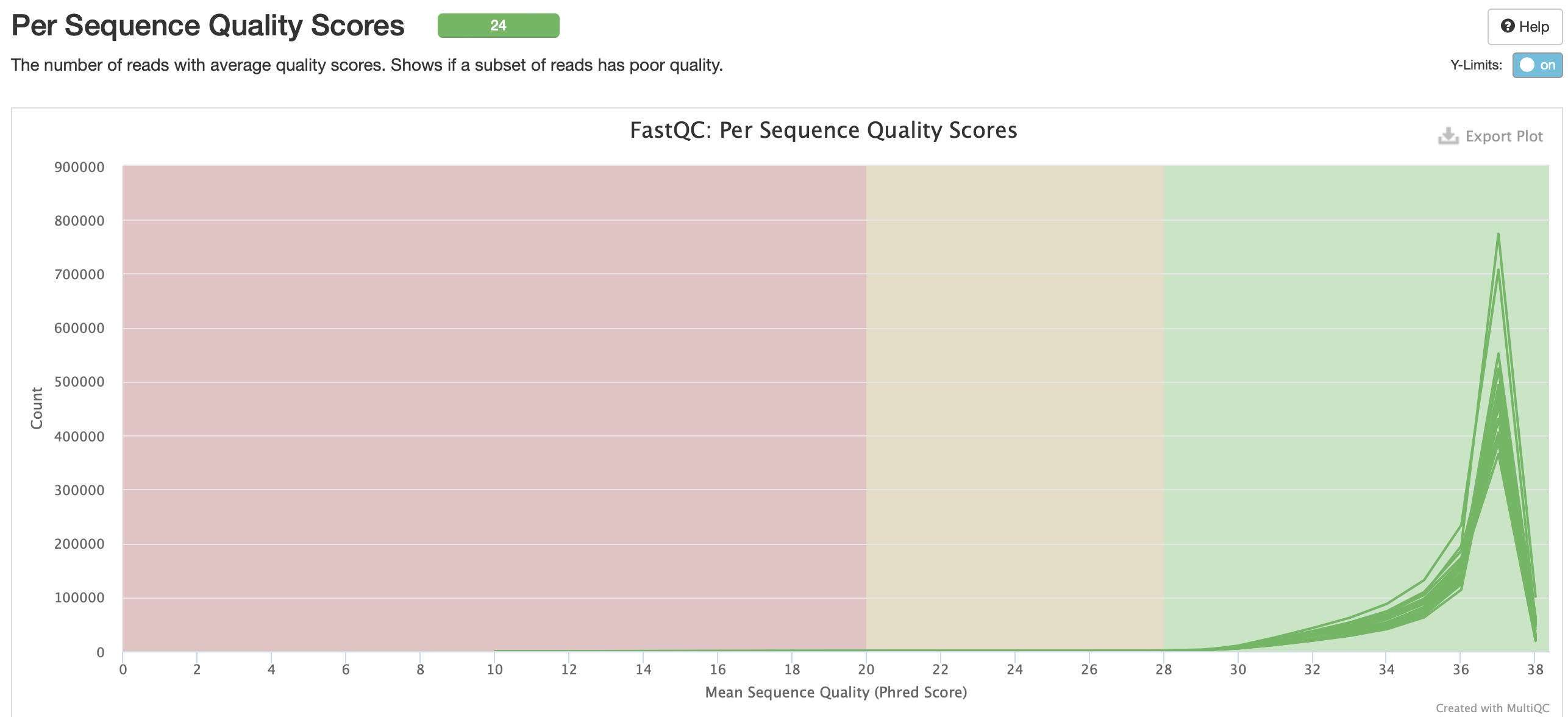
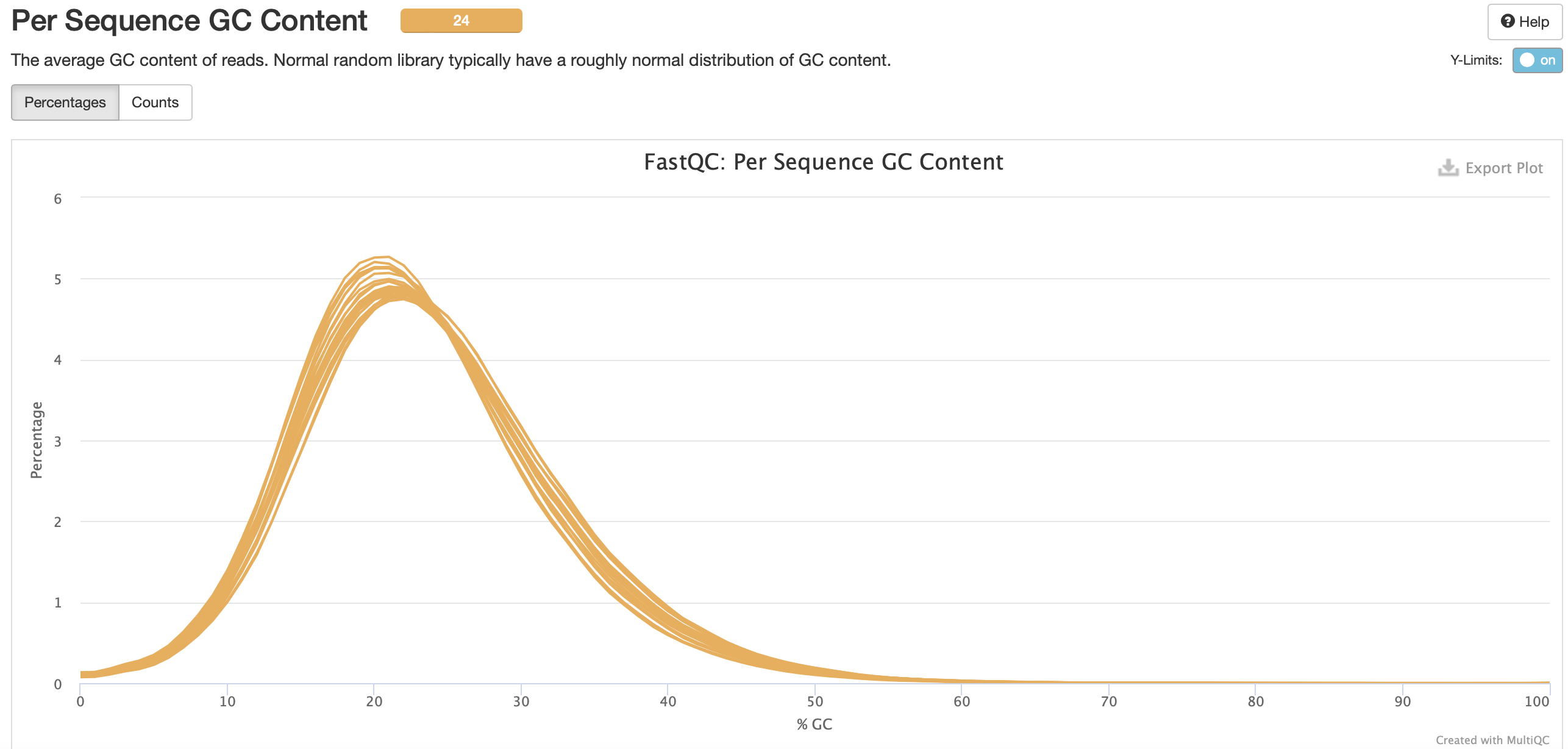
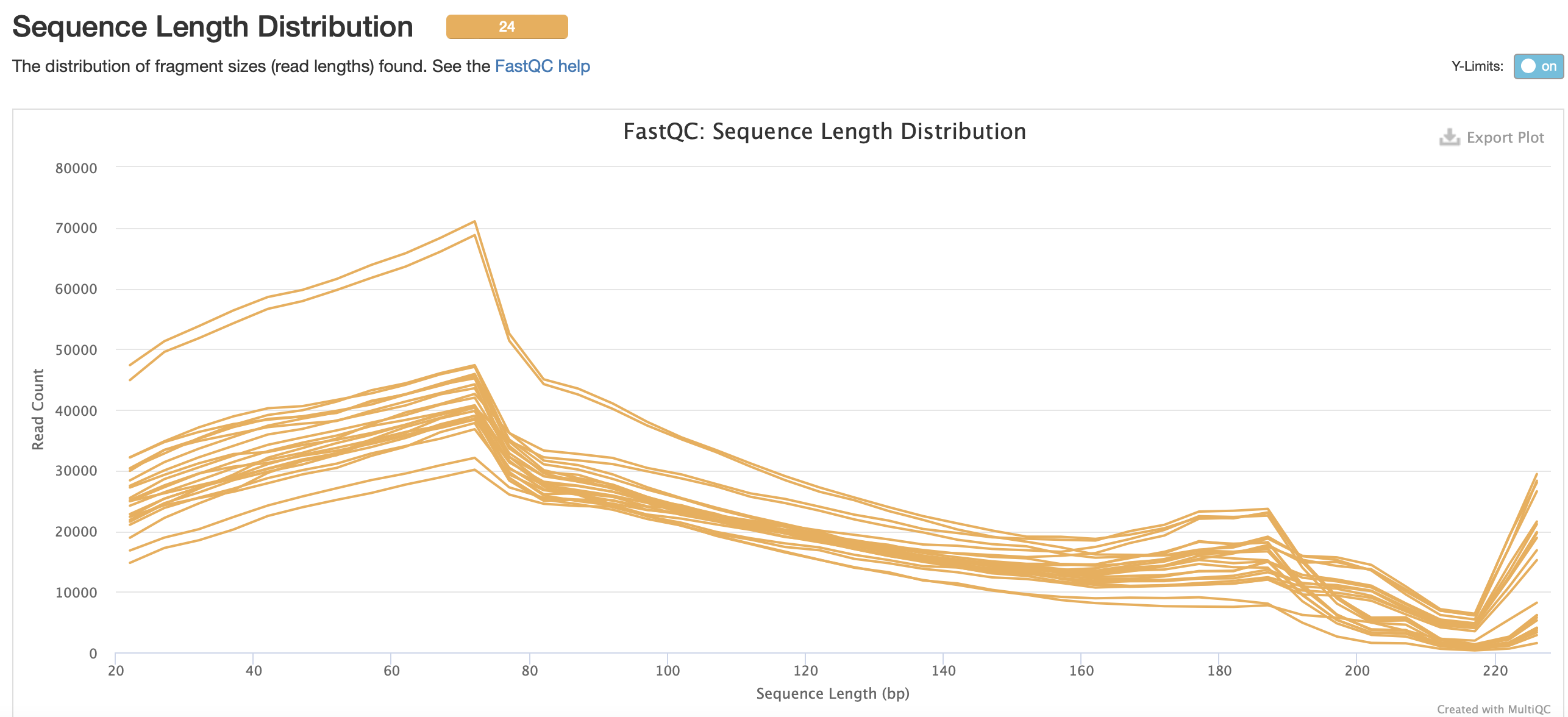
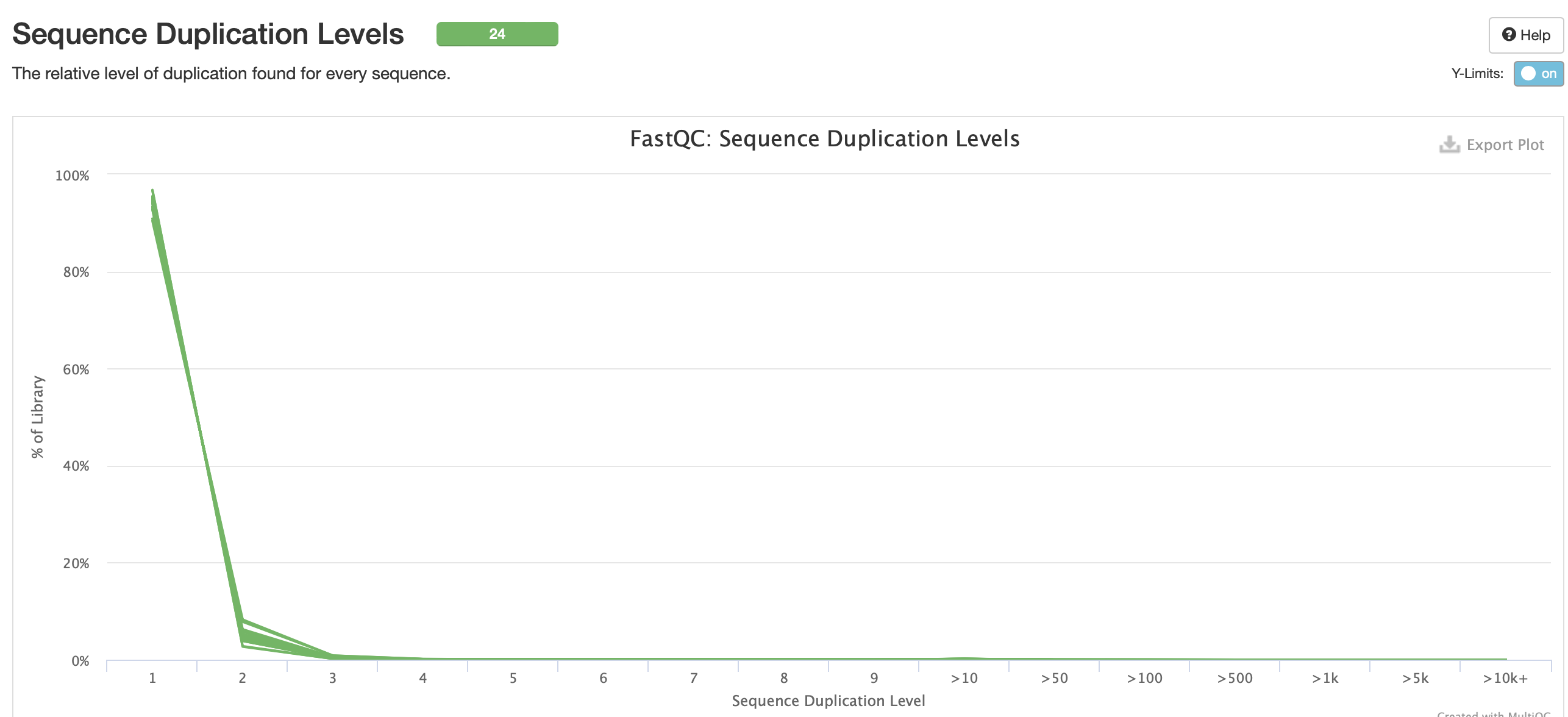
No adapter content present!
Based on the QC info, I’m going to continue with trim6 and bismark.
Bismark - iteration 2
I’m going to make a new directory for the 2nd iteration of Bismark. I’m also not going to make separate folders for the alignment/dedup info like last time because it seems like the downstream analysis may be easier with everything all in one folder.
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
mkdir bismark2
Prepare genome
I already prepped genome in a Bismark iteration above, so I don’t need to do it again. Can move on to aligning reads!
Align reads
Using the data from trim 6 iteration.
In scripts folder: nano bismark_align2.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_align2_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_align2_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load Bowtie2/2.4.4-GCC-11.2.0
echo "Starting Bismark alignment - 2nd iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim6
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
bismark --multicore 10 --bam --non_directional --output_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark2 --temp_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/temp --unmapped --ambiguous --genome /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs -1 ${file}_L001_R1_001_val_1.fq.gz -2 ${file}_L001_R2_001_val_2.fq.gz
done
echo "Bismark alignment complete! - 2nd iteration" $(date)
Still trying to decide if I should edit the --score_min argument. The default is --score_min L,0,-0.2. I’m going to run this script first and go from there. Submitted batch job 251643.
Deduplicate reads
In scripts folder: nano bismark_deduplicate2.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_deduplicate2_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_deduplicate2_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
echo "Starting Bismark deduplication - 2nd iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark2
for file in *bismark_bt2_pe.bam
do
deduplicate_bismark --paired --bam $file
done
echo "Bismark deduplication complete! - 2nd iteration" $(date)
Submitted batch job 251645
Sort reads??????
Extract methylation calls
Using the deduplicated.bam files
In scripts folder: nano bismark_methyl_extract2.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_methyl_extract2_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_methyl_extract2_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load SAMtools/1.16.1-GCC-11.3.0
echo "Starting to extract methylation calls - 2nd iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark2
for file in *deduplicated.bam
do
bismark_methylation_extractor --paired-end --bedGraph --scaffolds --cytosine_report --genome_folder /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs $file
done
echo "Methylation extraction complete - 2nd iteration!" $(date)
Submitted batch job 251646
QC / sample reports
In the above code, I ran bismark2report for each sample, but I don’t think I’m going to do that this time, as I didn’t really end up using/looking at those files. I can always come back to it if needed. I will run bismark2summary and MultiQC.
First, run bismark2summary code
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark2
module load Bismark/0.23.1-foss-2021b
bismark2summary *bismark_bt2_pe.bam
# scp summary to local computer
Now run MultiQC
module load MultiQC/1.9-intel-2020a-Python-3.8.2
multiqc -f --filename multiqc_report . \
-m custom_content -m picard -m qualimap -m bismark -m samtools -m preseq -m cutadapt -m fastqc
MultiQC results
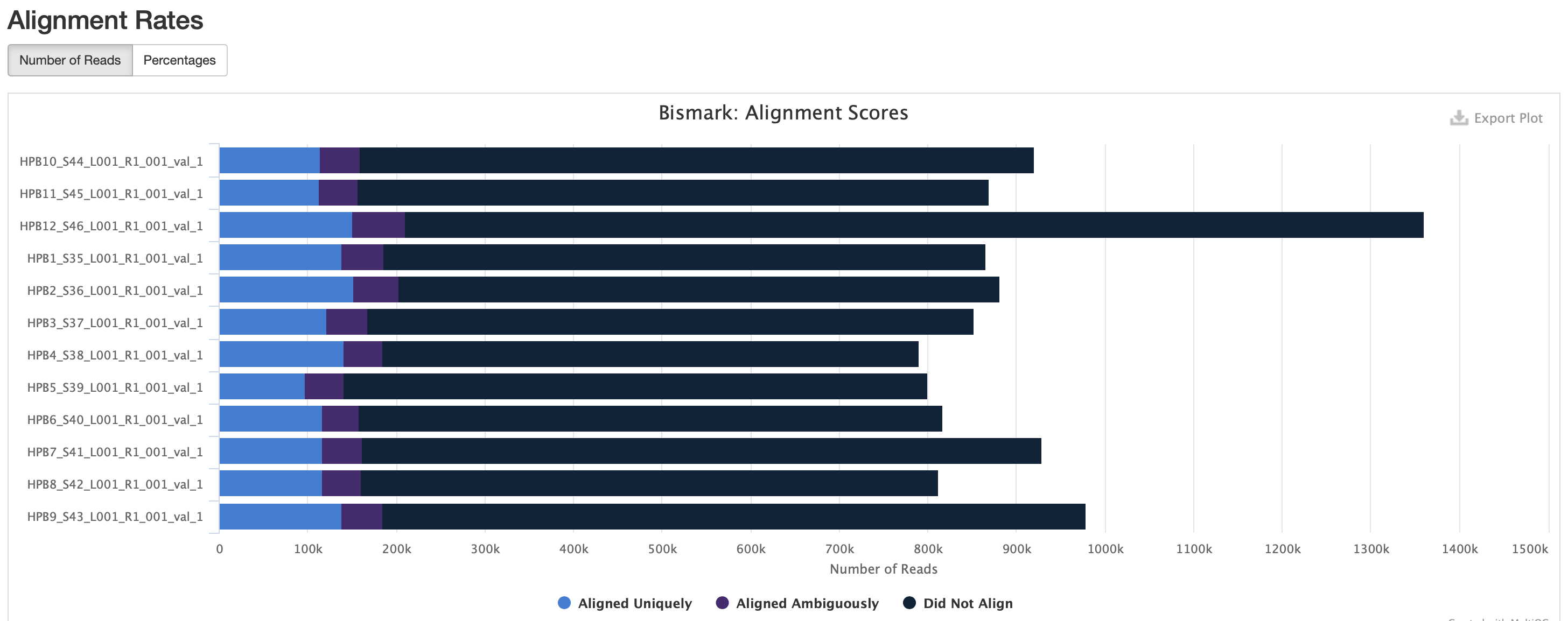
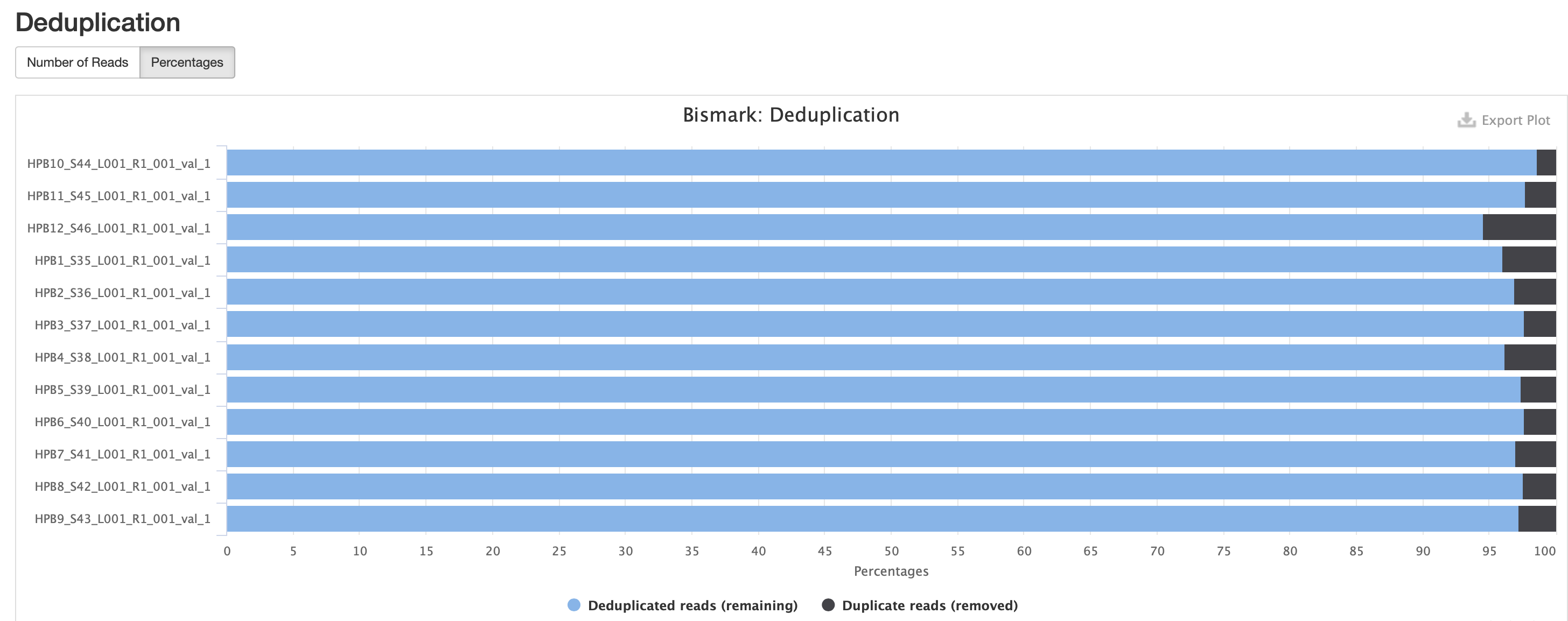
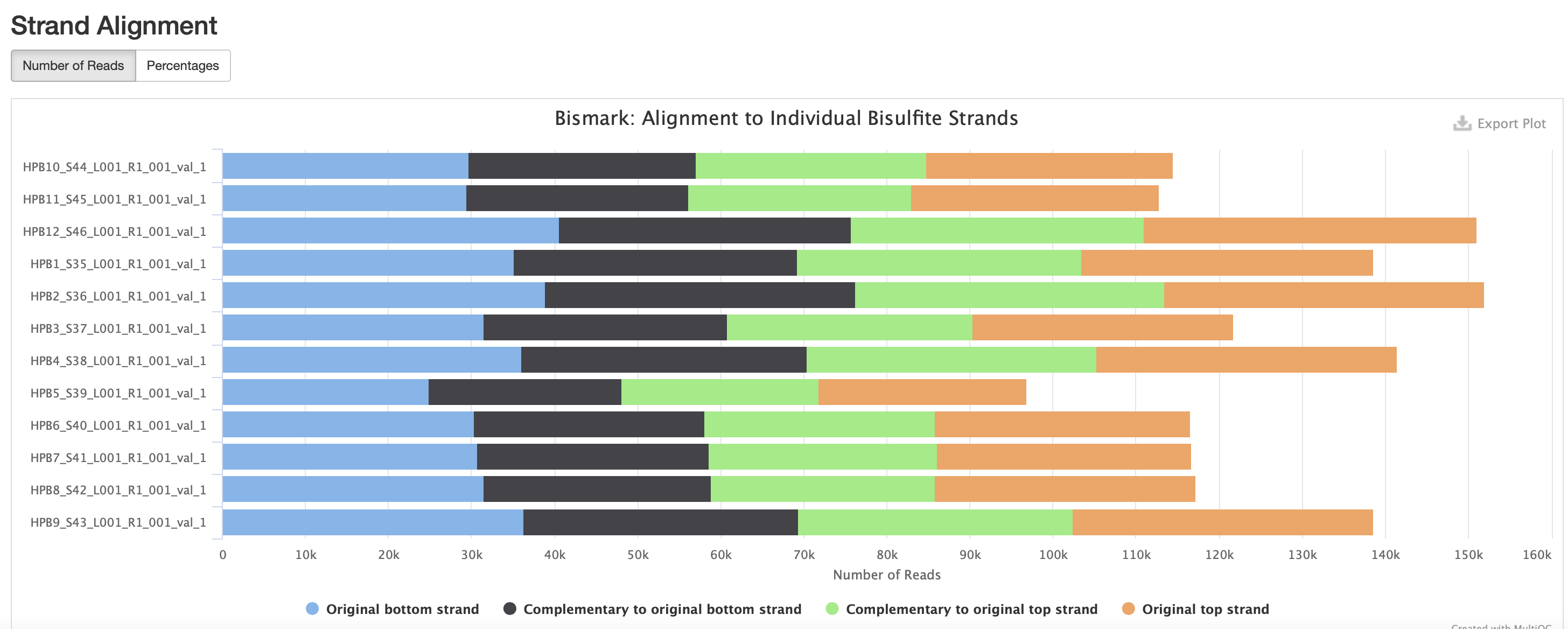
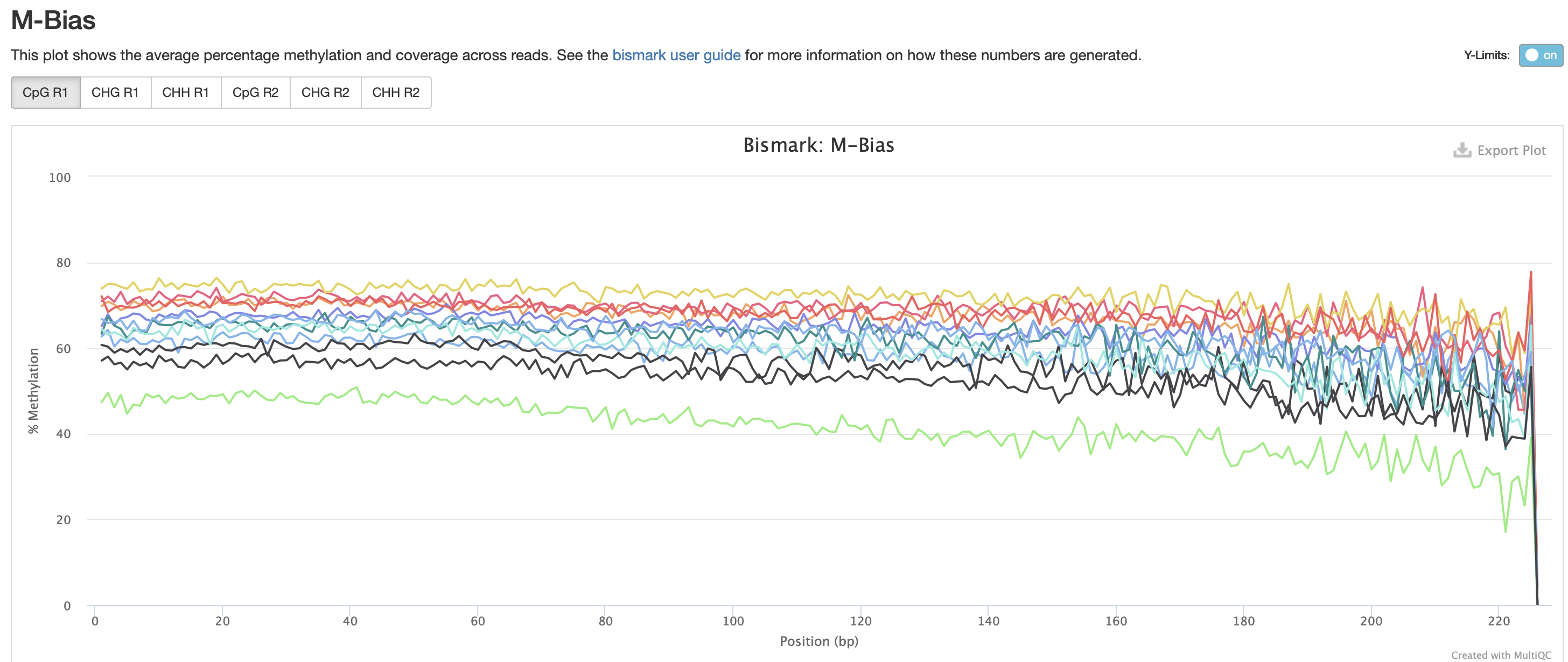
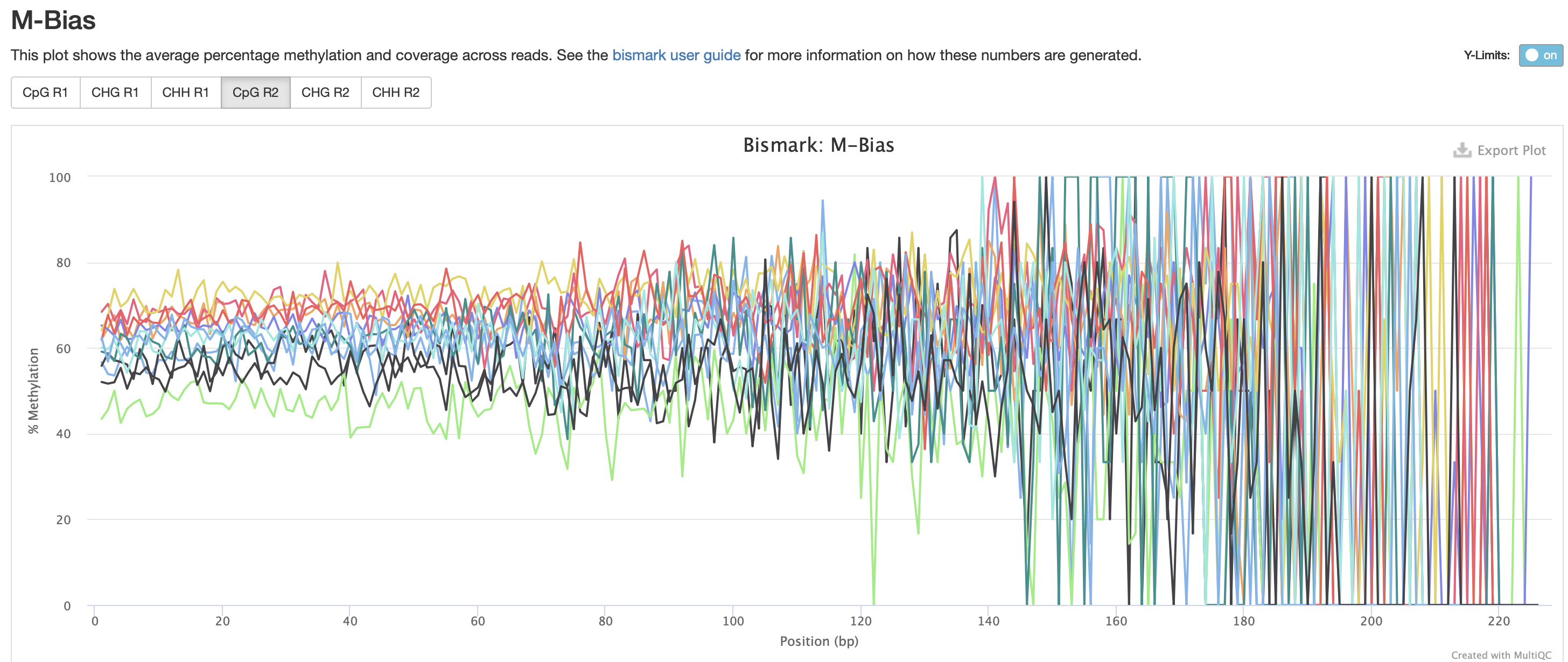
Okay M-bias is still all over the place. I’m going to try another iteration of Bismark with a less stringent alignment score.
Bismark - iteration 3
I’m going to make a new directory for the 3rd iteration of Bismark. I’m also not going to make separate folders for the alignment/dedup info like last time because it seems like the downstream analysis may be easier with everything all in one folder.
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
mkdir bismark3
I’ll be using trim6 data for this iteration.
Prepare genome
I already prepped genome in a Bismark iteration above, so I don’t need to do it again. Can move on to aligning reads!
Align reads
Using the data from trim 6 iteration.
In scripts folder: nano bismark_align3.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_align3_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_align3_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load Bowtie2/2.4.4-GCC-11.2.0
echo "Starting Bismark alignment - 3rd iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim6
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
bismark --multicore 10 --bam --non_directional --score_min L,0,-0.9 --output_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark3 --temp_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/temp --unmapped --ambiguous --genome /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs -1 ${file}_L001_R1_001_val_1.fq.gz -2 ${file}_L001_R2_001_val_2.fq.gz
done
echo "Bismark alignment complete! - 3rd iteration" $(date)
For this iteration, I changed the score min argument from -0.2 to -0.9. Hopefully, this will help with the M-bias that we’ve seen in the methylation data.
Submitted batch job 244196
Deduplicate reads
In scripts folder: nano bismark_deduplicate3.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_deduplicate3_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_deduplicate3_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
echo "Starting Bismark deduplication - 3rd iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark3
for file in *bismark_bt2_pe.bam
do
deduplicate_bismark --paired --bam $file
done
echo "Bismark deduplication complete! - 3rd iteration" $(date)
Submitted batch job 246166
Sort reads??????
Extract methylation calls
Using the deduplicated.bam files
In scripts folder: nano bismark_methyl_extract3.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_methyl_extract3_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_methyl_extract3_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load SAMtools/1.16.1-GCC-11.3.0
echo "Starting to extract methylation calls - 3rd iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark3
for file in *deduplicated.bam
do
bismark_methylation_extractor --paired-end --bedGraph --scaffolds --cytosine_report --genome_folder /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs $file
done
echo "Methylation extraction complete - 3rd iteration!" $(date)
Submitted batch job 246167
QC / sample reports
Above in bismark iteration 1, I ran bismark2report for each sample, but I don’t think I’m going to do that this time, as I didn’t really end up using/looking at those files. I can always come back to it if needed. I will run bismark2summary and MultiQC.
First, run bismark2summary code
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark3
module load Bismark/0.23.1-foss-2021b
bismark2summary *bismark_bt2_pe.bam
Now run MultiQC
module load MultiQC/1.9-intel-2020a-Python-3.8.2
multiqc -f --filename multiqc_report . \
-m custom_content -m picard -m qualimap -m bismark -m samtools -m preseq -m cutadapt -m fastqc
Secure copy files to local computer
MultiQC results
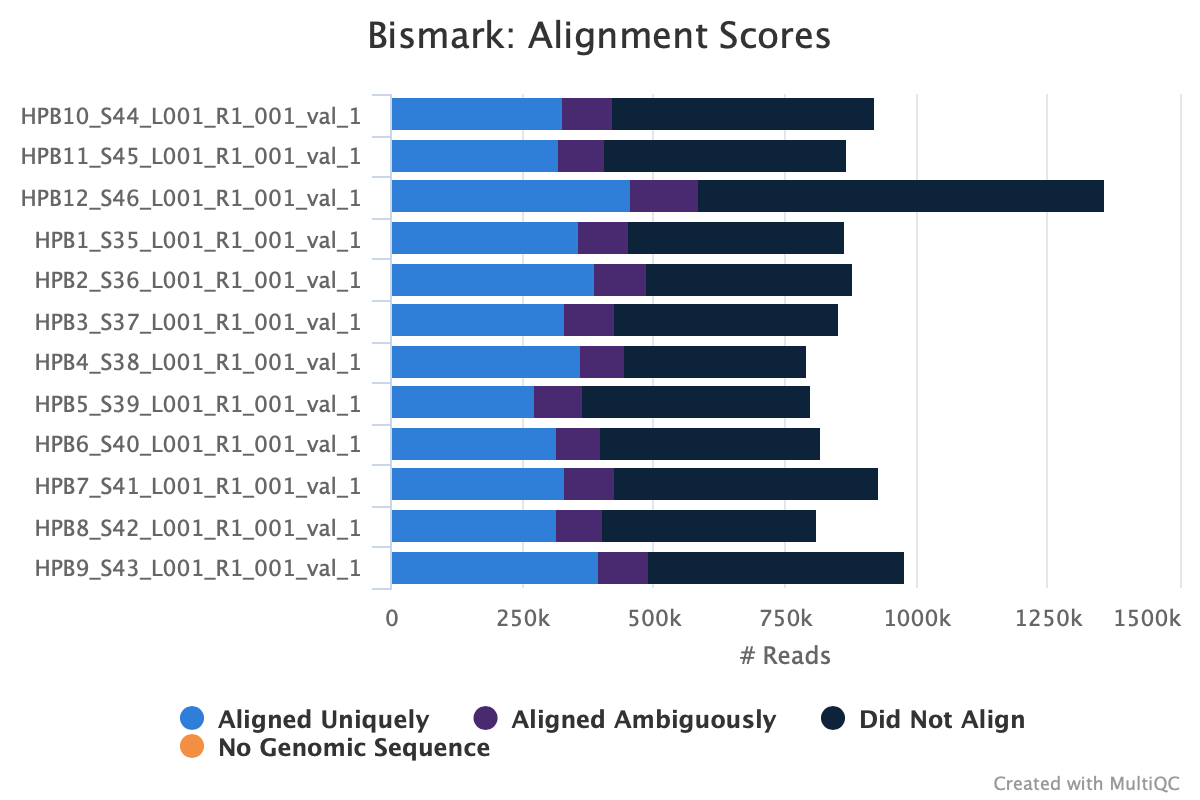
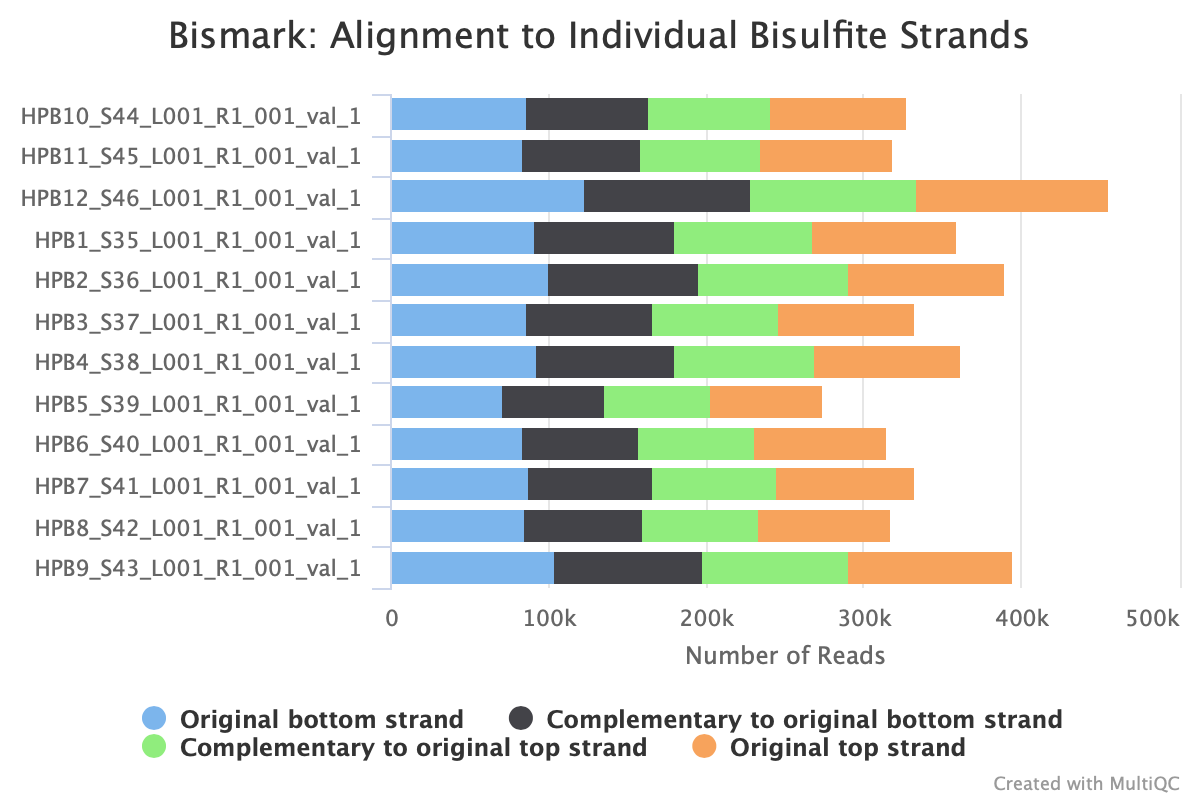
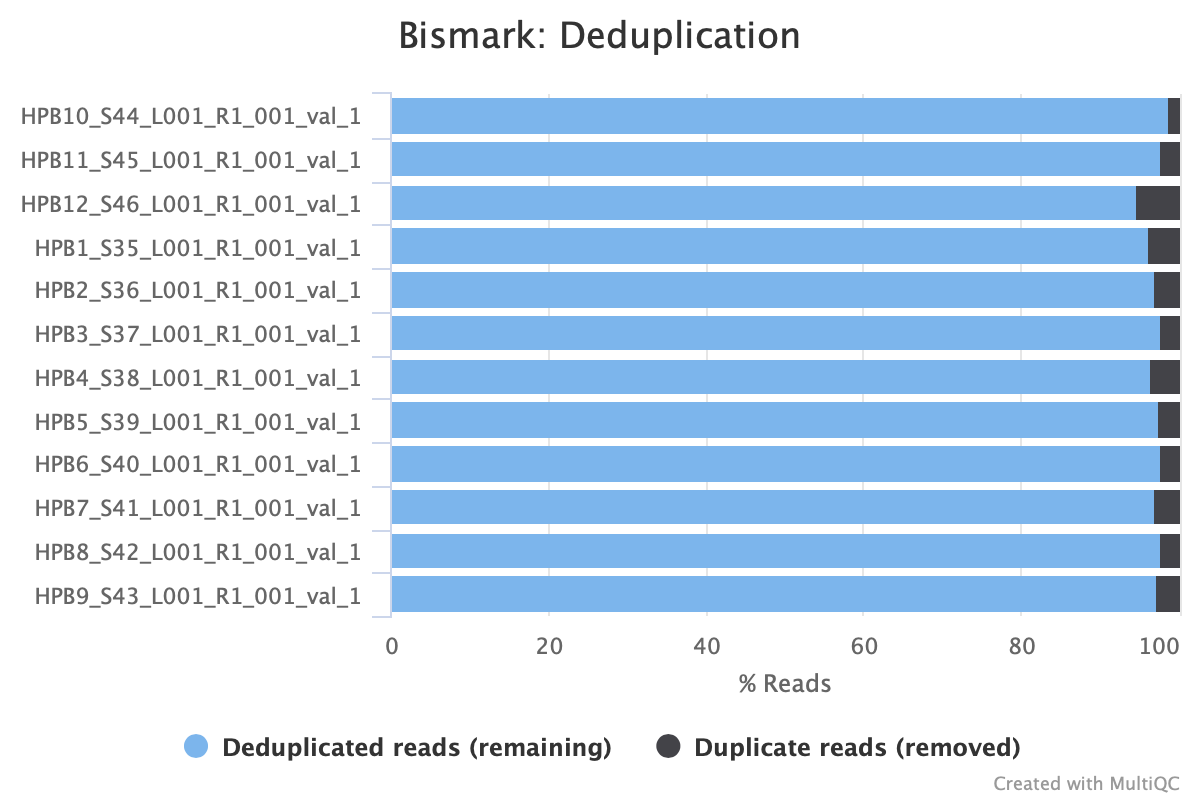
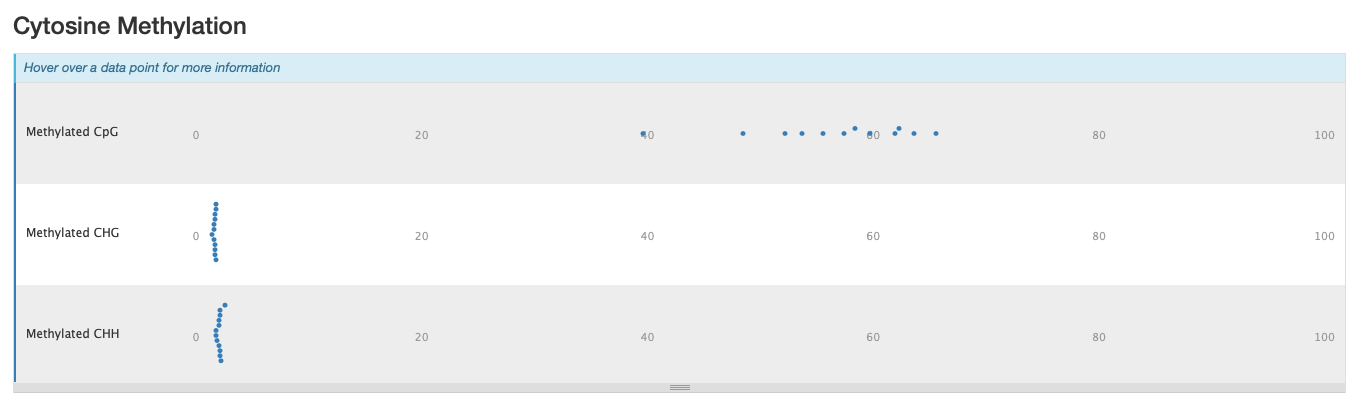
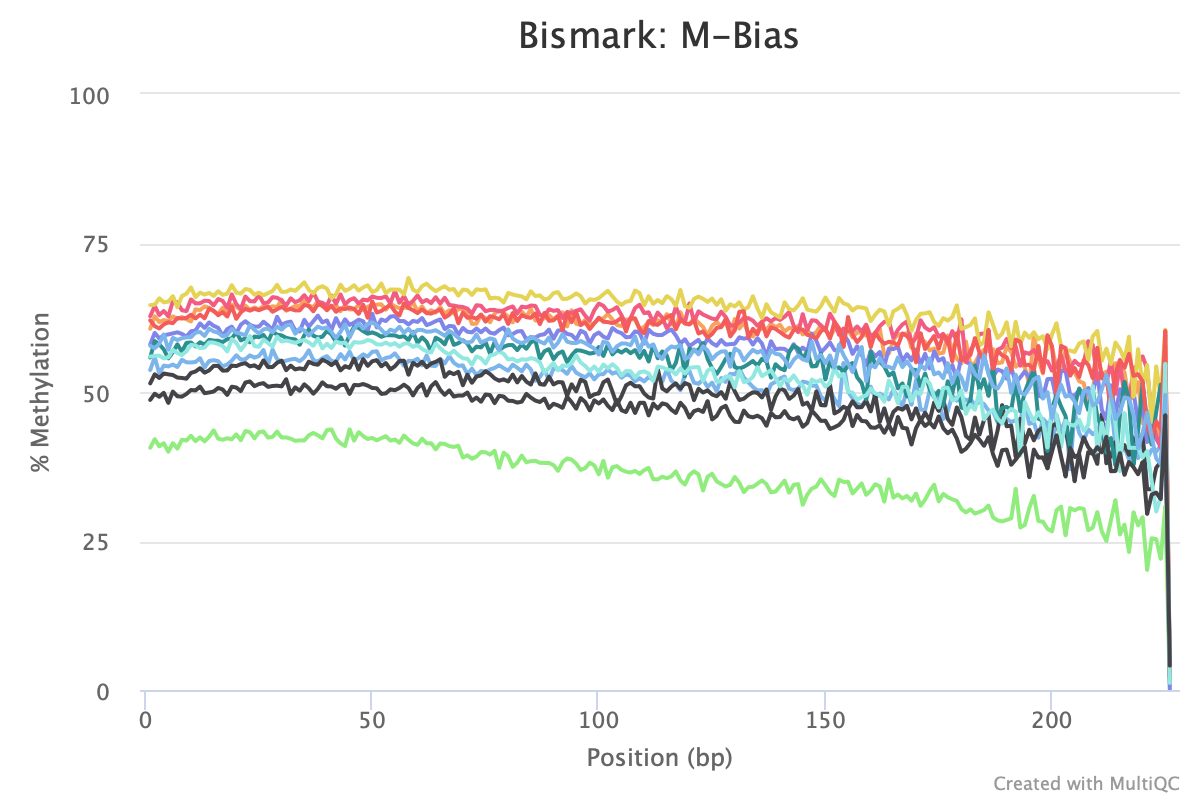
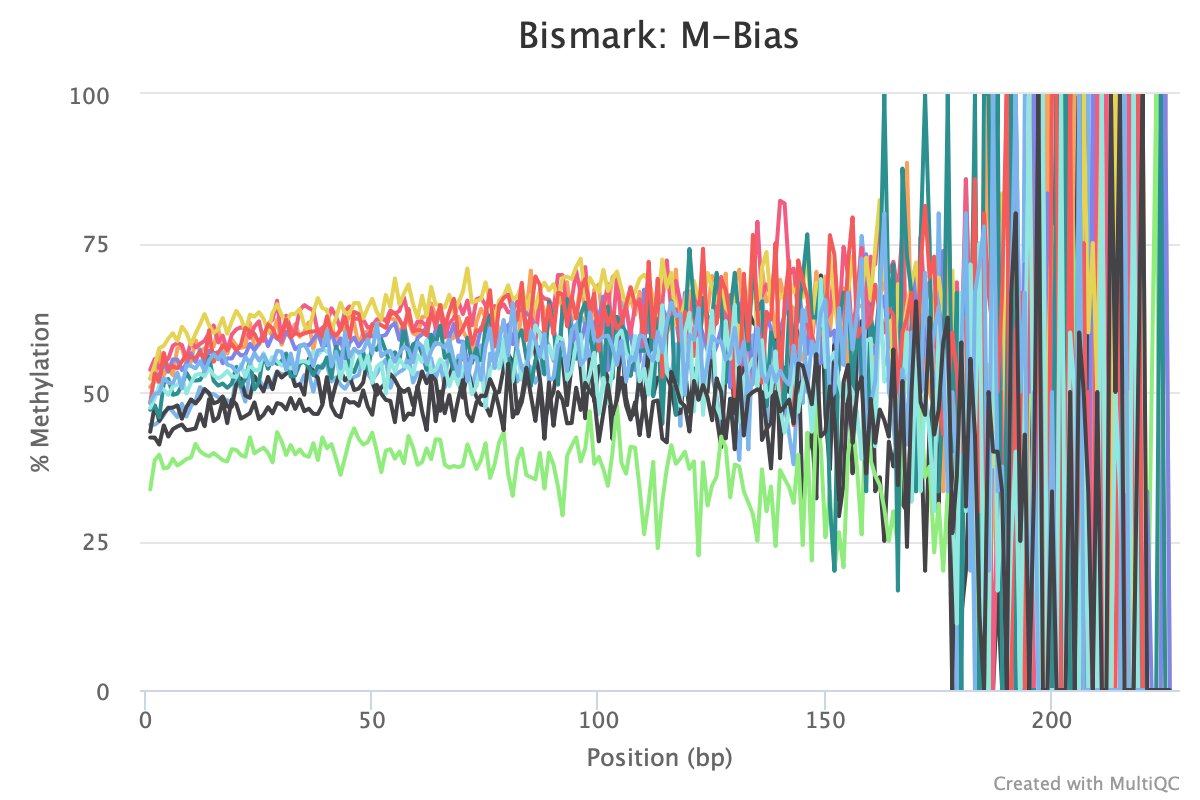
M-bias still all over the place. It starts to get pretty wonky around 150 bp, so let me try to cut 150 bp out. Going to try another trimming iteration in which I cut 99 bp and then 51 bp (I can only cut 99 bp at a time w/ trim galore).
Trim sequences
Make folders from trim7 iteration
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results
mkdir trim7
cd trim7
mkdir 99_bp_cut 150_bp_cut
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data
mkdir trim7
cd trim7
mkdir 99_bp_cut 150_bp_cut
Attempt 7
In scripts folder: nano trim_galore7.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="trim_galore7_error" #if your job fails, the error report will be put in this file
#SBATCH --output="trim_galore7_output" #once your job is completed, any final job report comments will be put in this file
module load Trim_Galore/0.6.7-GCCcore-11.2.0
echo "Trimming 150 bp from sequences - trim iteration 7" $(date)
# Cut 99 bp first
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/raw
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001.fastq.gz ${file}_L001_R2_001.fastq.gz --clip_r1 99 --clip_r2 99 --quality 30 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim7/99_bp_cut/
done
echo "Trimmed 99 bp from seqs so far" $(date)
# Cut another 51 bp
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim7/99_bp_cut
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
trim_galore --paired ${file}_L001_R1_001_val_1.fq.gz ${file}_L001_R1_001_val_2.fq.gz --clip_r1 51 --clip_r2 51 --quality 30 -o /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim7/150_bp_cut/
done
echo "Trimmed another 51 bp from seqs - trimming complete!" $(date)
Submitted batch job 246341
Trim sequences
Attempt 7
Going to run fastQC for both the 99 bp cut and the 150 bp cut
In scripts folder: nano fastqc_trim7.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="fastqc_trim7_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim7_output" #once your job is completed, any final job report comments will be put in this file
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
echo "QC for 99 bp cut" $(date)
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim7/99_bp_cut/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim7/99_bp_cut
done
multiqc --interactive fastqc_results/trim7/99_bp_cut
echo "QC for 150 bp cut" $(date)
for file in /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim7/150_bp_cut/*fq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/fastqc_results/trim7/150_bp_cut
done
multiqc --interactive fastqc_results/trim7/150_bp_cut
Submitted batch job 246394
Based on the QC info, I’m going to move forward with the analysis of 99 bp cut. The 150 bp cut looks like it totally erased the entirety of R2 for all samples.
Bismark - iteration 4
I’m going to make a new directory for the 4th iteration of Bismark. I’m also not going to make separate folders for the alignment/dedup info like last time because it seems like the downstream analysis may be easier with everything all in one folder.
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
mkdir bismark4
I’ll be using trim7 99 bp cut data for this iteration.
Prepare genome
I already prepped genome in a Bismark iteration above, so I don’t need to do it again. Can move on to aligning reads!
Align reads
Using the data from trim 6 iteration.
In scripts folder: nano bismark_align4.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_align4_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_align4_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load Bowtie2/2.4.4-GCC-11.2.0
echo "Starting Bismark alignment - 4th iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/data/trim7/99_bp_cut
for file in "HPB10_S44" "HPB11_S45" "HPB12_S46" "HPB1_S35" "HPB2_S36" "HPB3_S37" "HPB4_S38" "HPB5_S39" "HPB6_S40" "HPB7_S41" "HPB8_S42" "HPB9_S43"
do
bismark --multicore 10 --bam --non_directional --score_min L,0,-0.9 --output_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark4 --temp_dir /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark/temp --unmapped --ambiguous --genome /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs -1 ${file}_L001_R1_001_val_1.fq.gz -2 ${file}_L001_R2_001_val_2.fq.gz
done
echo "Bismark alignment complete! - 4th iteration" $(date)
For this iteration, I changed the score min argument from -0.2 to -0.9. Hopefully, this will help with the M-bias that we’ve seen in the methylation data. Submitted batch job 246395
Deduplicate reads
In scripts folder: nano bismark_deduplicate4.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_deduplicate4_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_deduplicate4_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
echo "Starting Bismark deduplication - 4th iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark4
for file in *bismark_bt2_pe.bam
do
deduplicate_bismark --paired --bam $file
done
echo "Bismark deduplication complete! - 4th iteration" $(date)
Submitted batch job 246397
Sort reads??????
Extract methylation calls
Using the deduplicated.bam files
In scripts folder: nano bismark_methyl_extract4.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_methyl_extract4_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_methyl_extract4_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load SAMtools/1.16.1-GCC-11.3.0
echo "Starting to extract methylation calls - 4th iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark4
for file in *deduplicated.bam
do
bismark_methylation_extractor --paired-end --bedGraph --scaffolds --cytosine_report --genome_folder /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs $file
done
echo "Methylation extraction complete - 4th iteration!" $(date)
Submitted batch job 246404
QC / sample reports
Above in bismark iteration 1, I ran bismark2report for each sample, but I don’t think I’m going to do that this time, as I didn’t really end up using/looking at those files. I can always come back to it if needed. I will run bismark2summary and MultiQC.
First, run bismark2summary code
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark4
module load Bismark/0.23.1-foss-2021b
bismark2summary *bismark_bt2_pe.bam
Now run MultiQC
module load MultiQC/1.9-intel-2020a-Python-3.8.2
multiqc -f --filename multiqc_report . \
-m custom_content -m picard -m qualimap -m bismark -m samtools -m preseq -m cutadapt -m fastqc
Secure copy files to local computer
MultiQC results
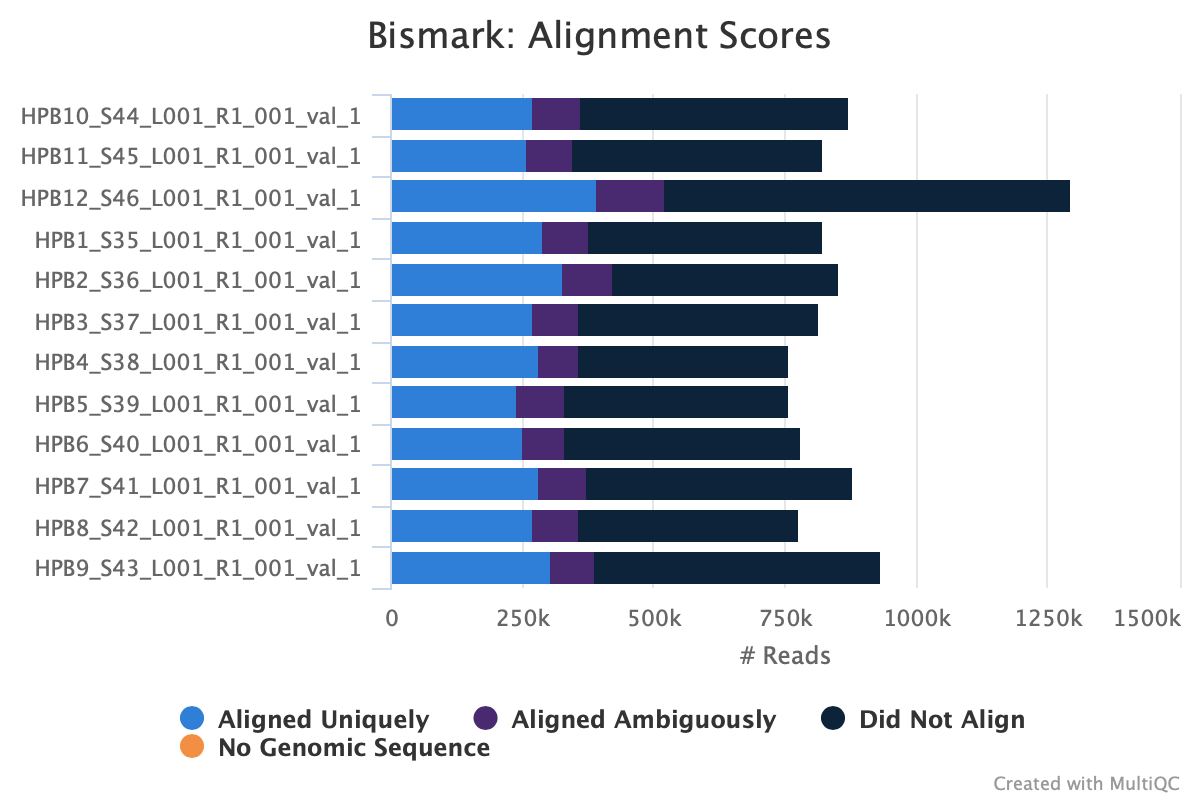
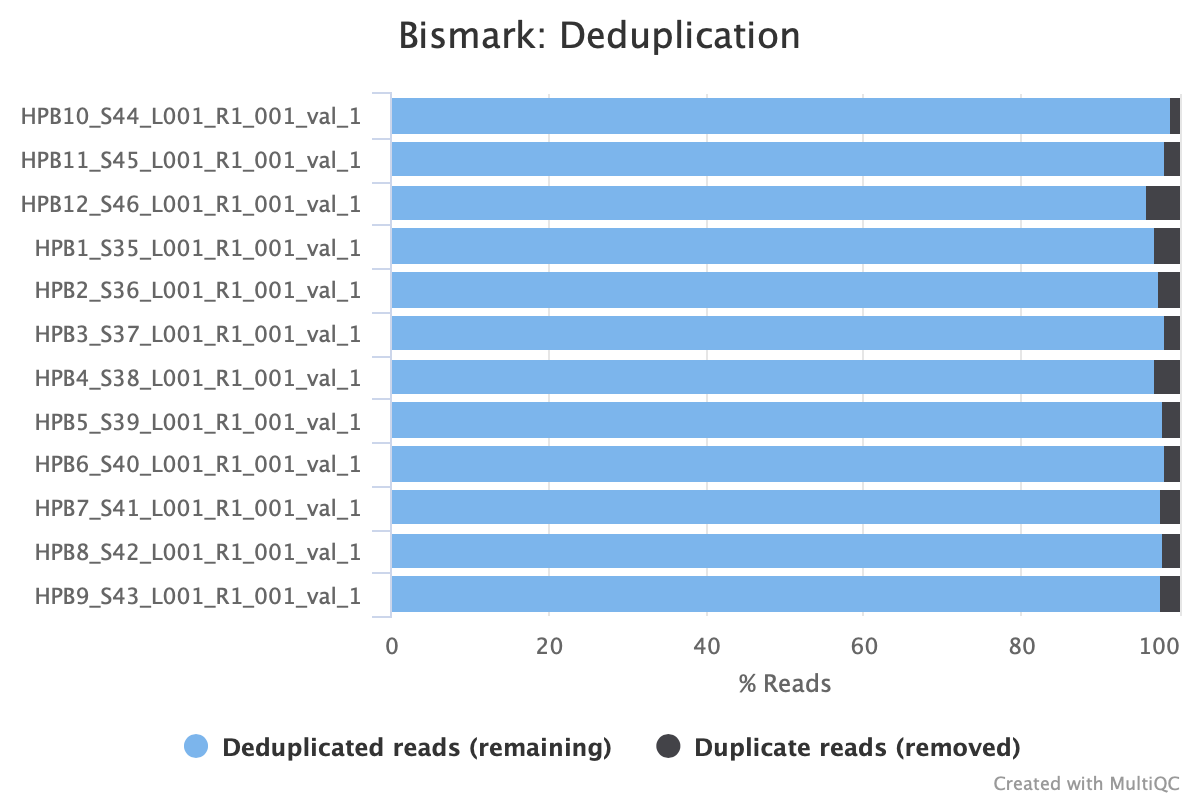
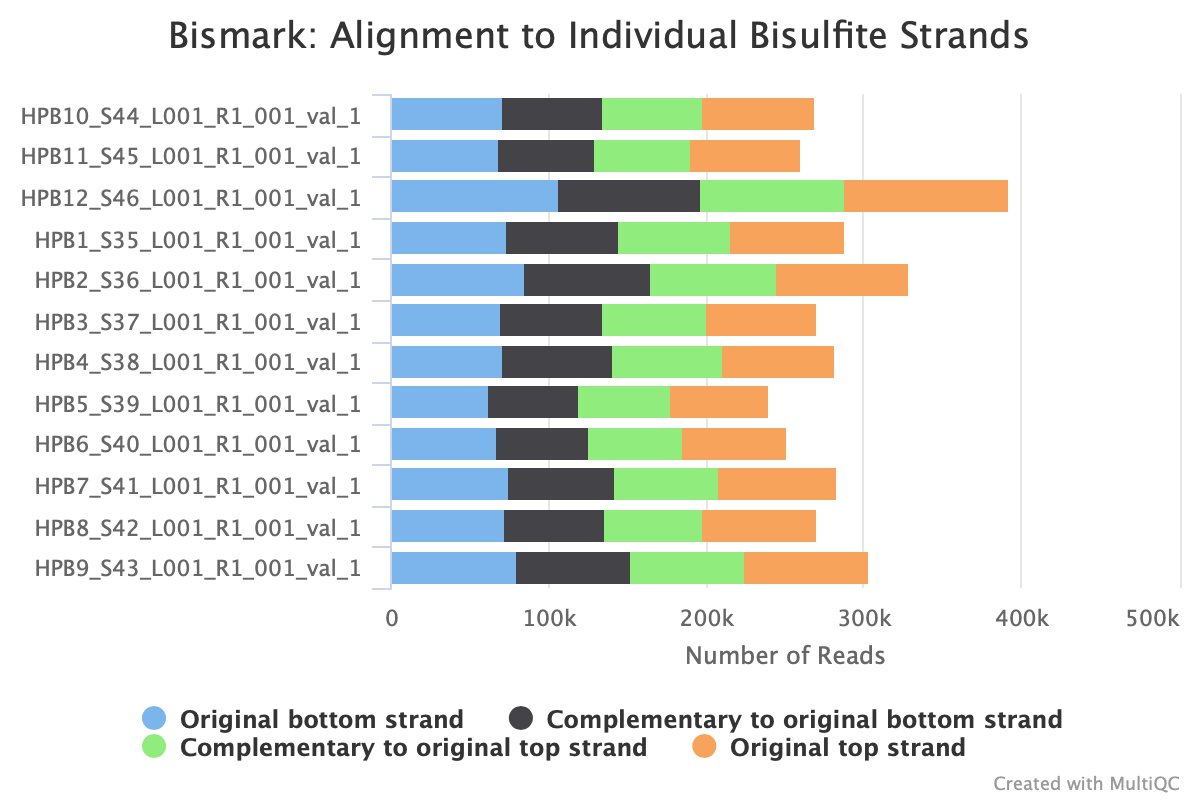
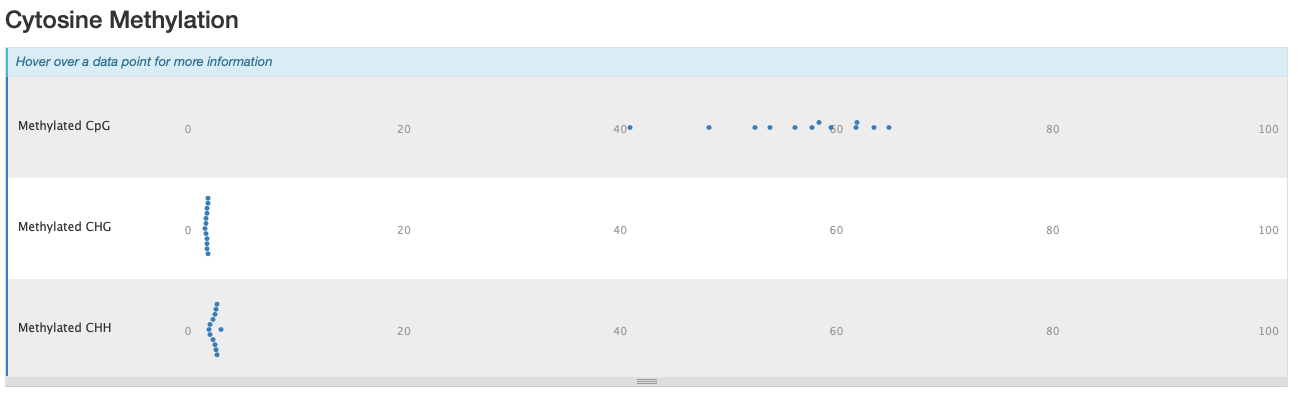
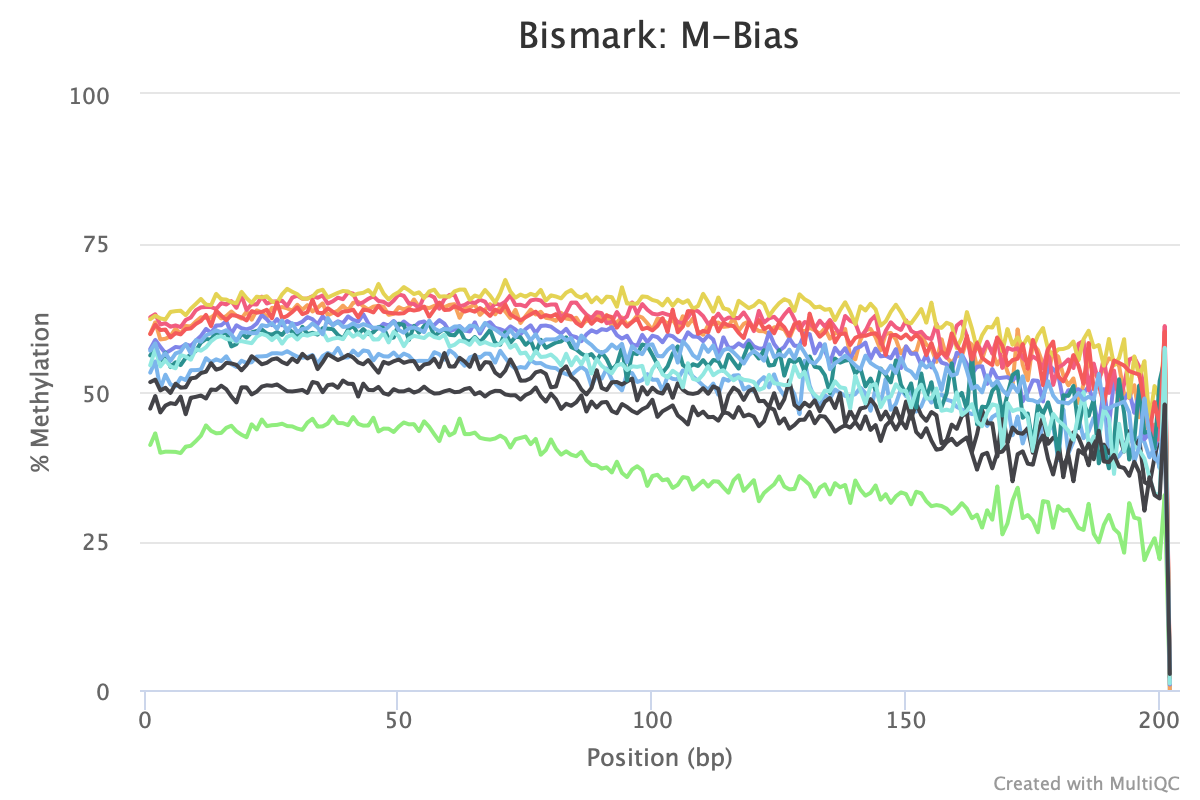
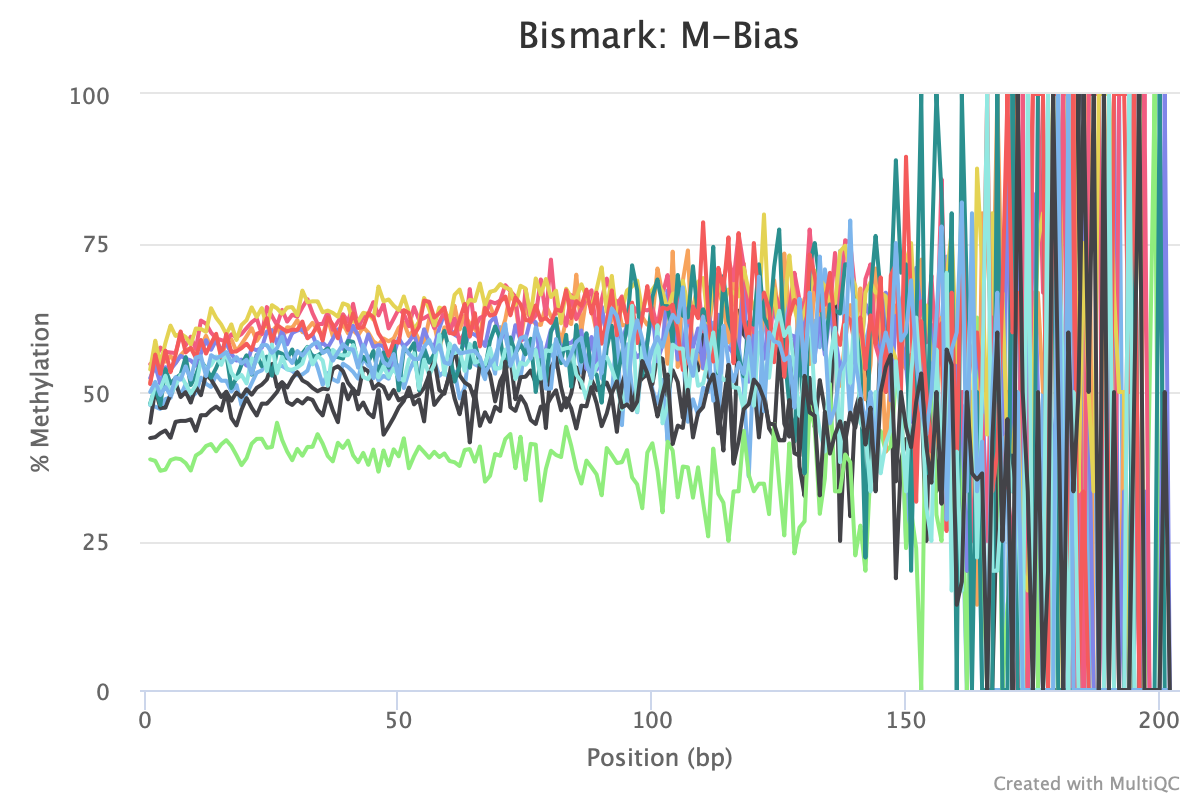
Still got some weird noise with the M-bias plot of Read 2. This post may provide insight on how to handle M-bias.
M-bias plot issues
Weird M-bias at the end of Read 2 has been present in all QC plots for each Bismark run. In the bismark_methylation_extractor command, there are options to ignore XX bases from the 5’ or 3’ end of Read 1 or Read 2. Lets try the code using the iteration4/trim7 information. Since we are starting from the extractor command, there is no need to rerun the alignment or deduplication commands.
Bismark - iteration 5
I’m going to make a new directory for the 5th iteration of Bismark. This is not really a brand new iteration, as I am using the data generated from iteration 4 (i.e., do not have to rerun the alignment or deduplication commands).
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS
mkdir bismark5
Extract methylation calls
Using the deduplicated.bam files in iteration 4 folder. I’m going to ignore 80 bp on the 3’ end of Read 2.
In scripts folder: nano bismark_methyl_extract5.sh
#!/bin/bash
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/scripts
#SBATCH --error="bismark_methyl_extract5_error" #if your job fails, the error report will be put in this file
#SBATCH --output="bismark_methyl_extract5_output" #once your job is completed, any final job report comments will be put in this file
module load Bismark/0.23.1-foss-2021b
module load SAMtools/1.16.1-GCC-11.3.0
echo "Starting to extract methylation calls - 5th iteration" $(date)
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark4
for file in *deduplicated.bam
do
bismark_methylation_extractor --paired-end --bedGraph --scaffolds --cytosine_report --output /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark5 --ignore_3prime_r2 80 --genome_folder /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/refs $file
done
echo "Methylation extraction complete - 5th iteration!" $(date)
Submitted batch job 262374
Copy bam and deduplicated bam files from bismark4 directory to bismark5 directory
cd /data/putnamlab/jillashey/Oys_Nutrient/MBDBS/bismark4
cp *val_1_bismark_bt2_pe.deduplicated.bam ../bismark5
cp *val_1_bismark_bt2_pe.bam ../bismark5
QC / sample reports
Run MultiQC
module load MultiQC/1.9-intel-2020a-Python-3.8.2
multiqc -f --filename multiqc_report . \
-m custom_content -m picard -m qualimap -m bismark -m samtools -m preseq -m cutadapt -m fastqc
Secure copy files to local computer
MultiQC results
Not giving me all the results… only see MultiQC graphs for cytosine methylation and M-bias. It does look like the M-bias 3’ ignore on Read 2 removed 80 bp, but its still pretty wonky towards the end.