
Astrangia 2021 small RNA analysis
Astrangia 2021 small RNA analysis
These data came from my Astrangia 2021 experiment, during which adult Astrangia colonies were exposed to ambient and high temperatures for ~9 months.
Files were downloaded to this location: /data/putnamlab/KITT/hputnam/20230605_Astrangia_smallRNA
Set-up
Make new folders for this project in my directory
cd /data/putnamlab/jillashey
mkdir Astrangia2021
cd Astrangia2021
mkdir smRNA
cd smRNA
mkdir data scripts fastqc
cd data
mkdir raw
cd raw
Copy the new files into raw data folder
cp /data/putnamlab/KITT/hputnam/20230605_Astrangia_smallRNA/* .
Check to make sure all files were transferred successfully
md5sum *.fastq.gz > checkmd5.md5
md5sum -c checkmd5.md5
AST-1065_R1_001.fastq.gz: OK
AST-1065_R2_001.fastq.gz: OK
AST-1105_R1_001.fastq.gz: OK
AST-1105_R2_001.fastq.gz: OK
AST-1147_R1_001.fastq.gz: OK
AST-1147_R2_001.fastq.gz: OK
AST-1412_R1_001.fastq.gz: OK
AST-1412_R2_001.fastq.gz: OK
AST-1560_R1_001.fastq.gz: OK
AST-1560_R2_001.fastq.gz: OK
AST-1567_R1_001.fastq.gz: OK
AST-1567_R2_001.fastq.gz: OK
AST-1617_R1_001.fastq.gz: OK
AST-1617_R2_001.fastq.gz: OK
AST-1722_R1_001.fastq.gz: OK
AST-1722_R2_001.fastq.gz: OK
AST-2000_R1_001.fastq.gz: OK
AST-2000_R2_001.fastq.gz: OK
AST-2007_R1_001.fastq.gz: OK
AST-2007_R2_001.fastq.gz: OK
AST-2302_R1_001.fastq.gz: OK
AST-2302_R2_001.fastq.gz: OK
AST-2360_R1_001.fastq.gz: OK
AST-2360_R2_001.fastq.gz: OK
AST-2398_R1_001.fastq.gz: OK
AST-2398_R2_001.fastq.gz: OK
AST-2404_R1_001.fastq.gz: OK
AST-2404_R2_001.fastq.gz: OK
AST-2412_R1_001.fastq.gz: OK
AST-2412_R2_001.fastq.gz: OK
AST-2512_R1_001.fastq.gz: OK
AST-2512_R2_001.fastq.gz: OK
AST-2523_R1_001.fastq.gz: OK
AST-2523_R2_001.fastq.gz: OK
AST-2563_R1_001.fastq.gz: OK
AST-2563_R2_001.fastq.gz: OK
AST-2729_R1_001.fastq.gz: OK
AST-2729_R2_001.fastq.gz: OK
AST-2755_R1_001.fastq.gz: OK
AST-2755_R2_001.fastq.gz: OK
Count number of reads per file
zgrep -c "@GWNJ" *fastq.gz
AST-1065_R1_001.fastq.gz:18782226
AST-1065_R2_001.fastq.gz:18782226
AST-1105_R1_001.fastq.gz:18535712
AST-1105_R2_001.fastq.gz:18535712
AST-1147_R1_001.fastq.gz:43815757
AST-1147_R2_001.fastq.gz:43815757
AST-1412_R1_001.fastq.gz:17729353
AST-1412_R2_001.fastq.gz:17729353
AST-1560_R1_001.fastq.gz:19958419
AST-1560_R2_001.fastq.gz:19958419
AST-1567_R1_001.fastq.gz:18414936
AST-1567_R2_001.fastq.gz:18414936
AST-1617_R1_001.fastq.gz:17164109
AST-1617_R2_001.fastq.gz:17164109
AST-1722_R1_001.fastq.gz:17993435
AST-1722_R2_001.fastq.gz:17993435
AST-2000_R1_001.fastq.gz:18885883
AST-2000_R2_001.fastq.gz:18885883
AST-2007_R1_001.fastq.gz:17958643
AST-2007_R2_001.fastq.gz:17958643
AST-2302_R1_001.fastq.gz:17901570
AST-2302_R2_001.fastq.gz:17901570
AST-2360_R1_001.fastq.gz:17996561
AST-2360_R2_001.fastq.gz:17996561
AST-2398_R1_001.fastq.gz:18231685
AST-2398_R2_001.fastq.gz:18231685
AST-2404_R1_001.fastq.gz:17661430
AST-2404_R2_001.fastq.gz:17661430
AST-2412_R1_001.fastq.gz:18215455
AST-2412_R2_001.fastq.gz:18215455
AST-2512_R1_001.fastq.gz:17643371
AST-2512_R2_001.fastq.gz:17643371
AST-2523_R1_001.fastq.gz:17901421
AST-2523_R2_001.fastq.gz:17901421
AST-2563_R1_001.fastq.gz:18067665
AST-2563_R2_001.fastq.gz:18067665
AST-2729_R1_001.fastq.gz:18840062
AST-2729_R2_001.fastq.gz:18840062
AST-2755_R1_001.fastq.gz:18122482
AST-2755_R2_001.fastq.gz:18122482
Raw QC
Run fastqc to quality check raw reads
In scripts folder: nano fastqc_raw.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH --error="fastqc_raw_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_raw_output" #once your job is completed, any final job report comments will be put in this file
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Astrangia2021/smRNA/fastqc/raw
done
multiqc --interactive fastqc_results
Submitted batch job 261246
Trim data
I have not done trimming specific to small RNAs, but this paper gave a nice workflow for miRNA analysis. They suggested using cutadapt. I’m going to follow their code, which applies a minimum length of 18 and a max length of 30. It doesn’t do any trimming of adapters, but we will see how the reads look after they go through this cutting.
In scripts folder: nano cutadapt.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH --error="cutadapt_error" #if your job fails, the error report will be put in this file
#SBATCH --output="cutadapt_output" #once your job is completed, any final job report comments will be put in this file
module load cutadapt/3.5-GCCcore-11.2.0
# Make array of sequences to cut
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
array1=($(ls *R1_001.fastq.gz))
echo "Trimming reads so that min length is 18 bp and max length is 30 bp" $(date)
# cutadapt loop
for i in ${array1[@]}; do
cutadapt --minimum-length=18 --maximum-length=30 -o /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/trimmed.${i} -p /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/trimmed.$(echo ${i}|sed s/_R1/_R2/) ${i} $(echo ${i}|sed s/_R1/_R2/)
done
echo "Trimming done!" $(date)
Submitted batch job 269180. Okay cutadapt not working. It is just cutting all of the reads because they are all long and the resulting trimmed file is just empty. Cancelling this job.
20230630 I should also ask Sam White/Javi about their trimming of miRNA data…
Questions for Javi/Sam
- details on seq for E5
- what trimming software did they use?
- did they do something like setting the min to 18 and max to 30
From Hao et al. 2021: “Raw reads obtained from the sequencing machine were filtered to get clean tags according to the following rules: removing low quality reads containing more than one low quality (Q-value≤ 20) base or containing unknown nucleotides(N) to get the high-quality reads. Then, high-quality reads were filtered by removing reads without 3′ adapters, containing 5′ adapters, containing 3′ and 5′ adapters but no small RNA fragment between them, containing polyA in small RNA fragment and shorter than 18 nt to get clean tags. The clean tags were aligned with small RNAs in the GenBank database”. This sounds like something i should try, but I’m not sure how to ID the 3’ and 5’ adapters in my sequences.
trimming - try cutadapt, trimmomatic or trimgalore
Looking at the adapter content MultiQC plot, it looks like the reads were processed using the illumina universal adapter and the illumina small rna 5’ adapter. The R1 reads have the universal adapter and the R2 reads have the small rna 5’ adapter. Not sure why that is. I looked the adapters sequences on Illumina and found this post that says the Illumina Universal Adapter—AGATCGGAAGAG and Illumina Small RNA 5’ Adapter—GATCGTCGGACT. I am unsure when this page wzs written, but I’m going to test them out.
nano cudadapt.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH --error="cutadapt_error" #if your job fails, the error report will be put in this file
#SBATCH --output="cutadapt_output" #once your job is completed, any final job report comments will be put in this file
module load cutadapt/3.5-GCCcore-11.2.0
# Make array of sequences to cut
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
array1=($(ls *R1_001.fastq.gz))
echo "Trimming reads so that min length is 18 bp and max length is 30 bp" $(date)
echo "Starting to trim using the Illumina universal adapter" $(date)
for i in ${array1[@]}; do
cutadapt -a AGATCGGAAGAG --minimum-length=18 --maximum-length=30 -o /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/trimmed.${i} -p /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/trimmed.$(echo ${i}|sed s/_R1/_R2/) ${i} $(echo ${i}|sed s/_R1/_R2/)
done
echo "Starting to trim using the Illumina small rna 5' adapter" $(date)
for i in ${array1[@]}; do
cutadapt -a GATCGTCGGACT --minimum-length=18 --maximum-length=30 -o /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/trimmed.again.${i} -p /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/trimmed.again.$(echo ${i}|sed s/_R1/_R2/) ${i} $(echo ${i}|sed s/_R1/_R2/)
done
echo "Trimming done!" $(date)
Submitted batch job 275252. The trimming parameters seem to be too stringent. It is saying that the reads are either too long or too short. Not sure what this means. I’m going to try Sam’s code where he trimmed some smRNAs using a software called flexbar. Need to ask Kevin Bryan to install flexbar.
Flexbar installed!
Let’s try Flexbar trimming code that Sam White wrote for the e5 small RNA analysis
First, I’m only going to try to trim one sample (2 reads) to see if flexbar works.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
First, make the NEB adapters fasta file.
nano NEB-adapters.fasta
>first
AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC
>second
GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATT
In scripts folder: nano test_flexbar.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="flexbar_raw_error" #if your job fails, the error report will be put in this file
#SBATCH --output="flexbar_raw_output" #once your job is completed, any final job report comments will be put in this file
module load Flexbar/3.5.0-foss-2018b
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
R1_fastq=/data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw/AST-1065_R1_001.fastq.gz
R2_fastq=/data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw/AST-1065_R2_001.fastq.gz
flexbar \
-r ${R1_fastq} \
-p ${R2_fastq} \
-a NEB-adapters.fasta \
-ap ON \
-qf i1.8 \
-qt 25 \
--post-trim-length 35 \
--target TEST_AST-1065 \
--zip-output GZ
Submitted batch job 284050. Finished in about 25 mins.
Now I’m going to run fastqc on the test sample and see how it looks:
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
fastqc TEST_AST-1065_1.fastq.gz TEST_AST-1065_2.fastq.gz
multiqc *fastqc*
The plots look good! I’m going to move forward w/ flexbar trimming.
In scripts folder: nano flexbar.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=200GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --error="flexbar_error" #if your job fails, the error report will be put in this file
#SBATCH --output="flexbar_output" #once your job is completed, any final job report comments will be put in this file
module load Flexbar/3.5.0-foss-2018b
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
echo "Trimming reads using flexbar" $(date)
array1=($(ls *R1_001.fastq.gz))
for i in ${array1[@]}; do
flexbar \
-r ${i} \
-p $(echo ${i}|sed s/_R1/_R2/) \
-a NEB-adapters.fasta \
-ap ON \
-qf i1.8 \
-qt 25 \
--post-trim-length 35 \
--target $(echo ${i}|sed s/_R1/_R2/) \
--zip-output GZ
done
Submitted batch job 284064
Move trimmed reads to trim folder
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
mv trim.AST-* ../trim/
Trim QC
Run fastqc to quality check trim reads
In scripts folder: nano fastqc_trim.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH --error="fastqc_trim_error" #if your job fails, the error report will be put in this file
#SBATCH --output="fastqc_trim_output" #once your job is completed, any final job report comments will be put in this file
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
for file in /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Astrangia2021/smRNA/fastqc/trim
done
multiqc --interactive fastqc_results/trim
Submitted batch job 284426
Count number of reads per file
zgrep -c "@GWNJ" *fastq.gz
trim.AST-1065_R2_001.fastq.gz_1.fastq.gz:17829111
trim.AST-1065_R2_001.fastq.gz_2.fastq.gz:17829111
trim.AST-1105_R2_001.fastq.gz_1.fastq.gz:17238126
trim.AST-1105_R2_001.fastq.gz_2.fastq.gz:17238126
trim.AST-1147_R2_001.fastq.gz_1.fastq.gz:40415224
trim.AST-1147_R2_001.fastq.gz_2.fastq.gz:40415224
trim.AST-1412_R2_001.fastq.gz_1.fastq.gz:16279555
trim.AST-1412_R2_001.fastq.gz_2.fastq.gz:16279555
trim.AST-1560_R2_001.fastq.gz_1.fastq.gz:17827024
trim.AST-1560_R2_001.fastq.gz_2.fastq.gz:17827024
trim.AST-1567_R2_001.fastq.gz_1.fastq.gz:16611397
trim.AST-1567_R2_001.fastq.gz_2.fastq.gz:16611397
trim.AST-1617_R2_001.fastq.gz_1.fastq.gz:16077717
trim.AST-1617_R2_001.fastq.gz_2.fastq.gz:16077717
trim.AST-1722_R2_001.fastq.gz_1.fastq.gz:16430221
trim.AST-1722_R2_001.fastq.gz_2.fastq.gz:16430221
trim.AST-2000_R2_001.fastq.gz_1.fastq.gz:17428854
trim.AST-2000_R2_001.fastq.gz_2.fastq.gz:17428854
trim.AST-2007_R2_001.fastq.gz_1.fastq.gz:16559551
trim.AST-2007_R2_001.fastq.gz_2.fastq.gz:16559551
trim.AST-2302_R2_001.fastq.gz_1.fastq.gz:16665370
trim.AST-2302_R2_001.fastq.gz_2.fastq.gz:16665370
trim.AST-2360_R2_001.fastq.gz_1.fastq.gz:16648356
trim.AST-2360_R2_001.fastq.gz_2.fastq.gz:16648356
trim.AST-2398_R2_001.fastq.gz_1.fastq.gz:16788208
trim.AST-2398_R2_001.fastq.gz_2.fastq.gz:16788208
trim.AST-2404_R2_001.fastq.gz_1.fastq.gz:16712903
trim.AST-2404_R2_001.fastq.gz_2.fastq.gz:16712903
trim.AST-2412_R2_001.fastq.gz_1.fastq.gz:17488508
trim.AST-2412_R2_001.fastq.gz_2.fastq.gz:17488508
trim.AST-2512_R2_001.fastq.gz_1.fastq.gz:16265716
trim.AST-2512_R2_001.fastq.gz_2.fastq.gz:16265716
trim.AST-2523_R2_001.fastq.gz_1.fastq.gz:16995265
trim.AST-2523_R2_001.fastq.gz_2.fastq.gz:16995265
trim.AST-2563_R2_001.fastq.gz_1.fastq.gz:17023002
trim.AST-2563_R2_001.fastq.gz_2.fastq.gz:17023002
trim.AST-2729_R2_001.fastq.gz_1.fastq.gz:17121869
trim.AST-2729_R2_001.fastq.gz_2.fastq.gz:17121869
trim.AST-2755_R2_001.fastq.gz_1.fastq.gz:16269823
trim.AST-2755_R2_001.fastq.gz_2.fastq.gz:16269823
20231130
Locations of cnidarian miRNA data
- Stylophora pistillata
- Hydra
- Nematostella
- Acropora muricata, Montipora capricornis, Montipora foliosa, Pocillopora verrucosa
20240103 Should I retrim with fastp to keep it consistent with mRNA? Going to try it out and compare results from flexbar. In trimmed smRNA folder, make new directory to put fastp trimmed reads
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim
mkdir fastp
In scripts folder: nano fastp_QC.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.out
# Load modules needed
module load fastp/0.19.7-foss-2018b
module load FastQC/0.11.8-Java-1.8
module load MultiQC/1.9-intel-2020a-Python-3.8.2
# Make an array of sequences to trim in raw data directory
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
array1=($(ls *R1_001.fastq.gz))
echo "Read trimming of adapters started." $(date)
# fastp and fastqc loop
for i in ${array1[@]}; do
fastp --in1 ${i} \
--in2 $(echo ${i}|sed s/_R1/_R2/)\
--out1 /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/trimmed.${i} \
--out2 /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/trimmed.$(echo ${i}|sed s/_R1/_R2/) \
--detect_adapter_for_pe \
--qualified_quality_phred 30 \
--unqualified_percent_limit 10 \
#--length_required 100 \
--cut_right cut_right_window_size 5 cut_right_mean_quality 20
fastqc /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/trimmed.${i}
fastqc /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/trimmed.$(echo ${i}|sed s/_R1/_R2/)
done
echo "Read trimming of adapters complete." $(date)
# Quality Assessment of Trimmed Reads
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp #go to output directory
# Compile MultiQC report from FastQC files
multiqc --interactive ./
echo "Cleaned MultiQC report generated." $(date)
Submitted batch job 291969. Took about 5 hours. The QC plots don’t look amazing and the length is still at 100 bp. I’m going to rerun but adding the argument --length_limit 30 for fastp. This means that reads longer than 30 bp will be discarded. Submitted batch job 291997. Downloaded the QC report and it looks okay. Low amount of reads but high duplication. Let’s see how many counts are in each file:
zgrep -c "@GWNJ" *fastq.gz
trimmed.AST-1065_R1_001.fastq.gz:7834561
trimmed.AST-1065_R2_001.fastq.gz:7834561
trimmed.AST-1105_R1_001.fastq.gz:8754651
trimmed.AST-1105_R2_001.fastq.gz:8754651
trimmed.AST-1147_R1_001.fastq.gz:22326820
trimmed.AST-1147_R2_001.fastq.gz:22326820
trimmed.AST-1412_R1_001.fastq.gz:8158050
trimmed.AST-1412_R2_001.fastq.gz:8158050
trimmed.AST-1560_R1_001.fastq.gz:8733402
trimmed.AST-1560_R2_001.fastq.gz:8733402
trimmed.AST-1567_R1_001.fastq.gz:9830273
trimmed.AST-1567_R2_001.fastq.gz:9830273
trimmed.AST-1617_R1_001.fastq.gz:8146294
trimmed.AST-1617_R2_001.fastq.gz:8146294
trimmed.AST-1722_R1_001.fastq.gz:9014021
trimmed.AST-1722_R2_001.fastq.gz:9014021
trimmed.AST-2000_R1_001.fastq.gz:10252309
trimmed.AST-2000_R2_001.fastq.gz:10252309
trimmed.AST-2007_R1_001.fastq.gz:9622779
trimmed.AST-2007_R2_001.fastq.gz:9622779
trimmed.AST-2302_R1_001.fastq.gz:8921101
trimmed.AST-2302_R2_001.fastq.gz:8921101
trimmed.AST-2360_R1_001.fastq.gz:8635502
trimmed.AST-2360_R2_001.fastq.gz:8635502
trimmed.AST-2398_R1_001.fastq.gz:9565008
trimmed.AST-2398_R2_001.fastq.gz:9565008
trimmed.AST-2404_R1_001.fastq.gz:8798584
trimmed.AST-2404_R2_001.fastq.gz:8798584
trimmed.AST-2412_R1_001.fastq.gz:7032530
trimmed.AST-2412_R2_001.fastq.gz:7032530
trimmed.AST-2512_R1_001.fastq.gz:7463762
trimmed.AST-2512_R2_001.fastq.gz:7463762
trimmed.AST-2523_R1_001.fastq.gz:8272150
trimmed.AST-2523_R2_001.fastq.gz:8272150
trimmed.AST-2563_R1_001.fastq.gz:8537237
trimmed.AST-2563_R2_001.fastq.gz:8537237
trimmed.AST-2729_R1_001.fastq.gz:7929477
trimmed.AST-2729_R2_001.fastq.gz:7929477
trimmed.AST-2755_R1_001.fastq.gz:9775688
trimmed.AST-2755_R2_001.fastq.gz:9775688
Flexbar keeps more reads but it seems like it is combining the two reads into one for each sample (which I don’t want to do yet). Let’s edit the flexbar code and see if I can fix that. For now, I just commented out the line --target trim.$(echo ${i}|sed s/_R1/_R2/) \, which names the files. Also changing max read length to 30. Submitted batch job 292029
20240104
Flexbar trimming finished last night but it just rewrote the files over one another so only the last sample has the files. Looking at the flexbar documentation, it seems like --target is the prefix for output file names or paths, where as --output-reads and --output-reads2 is used for the output file for reads 1 and 2 instead of the target prefix usage. So I am just dumb and should’ve specified the output reads argument instead of target.
Let’s look at the flexbar script again:
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=200GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Flexbar/3.5.0-foss-2018b
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
echo "Trimming reads using flexbar" $(date)
array1=($(ls *R1_001.fastq.gz))
for i in ${array1[@]}; do
flexbar \
-r ${i} \
-p $(echo ${i}|sed s/_R1/_R2/) \
-a NEB-adapters.fasta \
-ap ON \
-qf i1.8 \
-qt 30 \
-t /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
--post-trim-length 30 \
--output-reads trim.${i} \
--output-reads2 trim.$(echo ${i}|sed s/_R1/_R2/) \
--zip-output GZ
done
Made those changes for output reads to the script. Also changed script so that -qt was 30 (ie phred score of 30) and post trim length was 30 bp. Submitted batch job 292075. Kept failing, for some reason all my files were empty. I’m now re-copying them from the KITT directory (thank god for backups) and then will rerun flexbar. Added trim. prefix to the beginning of the output file names so that the original files don’t get overwritten. Submitted batch job 292079.
Checked back and it is still overwriting the files with the flexbar.log and flexbar fastq files…Also in the slurm error report, it tells me that the post trim length argument was not found. Looking at the code again, I didn’t add a \ after the -t line. Going to add it and rerun. Also going to try to take out the -- and just do the -. Submitted batch job 292092
Job finished but the files are empty. Why does Flexbar hate me? The error file said it couldn’t open the files that flexbar created but why did those need to be opened anyway?
I’m going to go back to the original script that I ran because it seems to have worked, even though it named both files R2.
In the scripts folder: nano flexbar_og.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=200GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Flexbar/3.5.0-foss-2018b
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
echo "Trimming reads using flexbar" $(date)
array1=($(ls *R1_001.fastq.gz))
for i in ${array1[@]}; do
flexbar \
-r ${i} \
-p $(echo ${i}|sed s/_R1/_R2/) \
-a NEB-adapters.fasta \
-ap ON \
-qf i1.8 \
-qt 30 \
--post-trim-length 30 \
--target ${i} \
--zip-output GZ
done
Submitted batch job 292095
While that is running, I’m going to figure out how to set up a conda environment using the conda unity documentation. For whatever reason, Kevin Bryan recommended I use a conda environment.
I’m going to make it in the putnam lab folder.
First I need to load miniconda module: module load Miniconda3/4.9.2
Now I need to create a conda environment.
conda create --prefix /data/putnamlab/miranda
If I need to update:
==> WARNING: A newer version of conda exists. <==
current version: 4.9.2
latest version: 23.11.0
Please update conda by running
$ conda update -n base -c defaults conda
Now that the conda environment is created, I need to activate it.
conda activate /data/putnamlab/miranda
It told me my shell had not been properly configured. To do that:
conda init
no change /opt/software/Miniconda3/4.9.2/condabin/conda
no change /opt/software/Miniconda3/4.9.2/bin/conda
no change /opt/software/Miniconda3/4.9.2/bin/conda-env
no change /opt/software/Miniconda3/4.9.2/bin/activate
no change /opt/software/Miniconda3/4.9.2/bin/deactivate
no change /opt/software/Miniconda3/4.9.2/etc/profile.d/conda.sh
no change /opt/software/Miniconda3/4.9.2/etc/fish/conf.d/conda.fish
no change /opt/software/Miniconda3/4.9.2/shell/condabin/Conda.psm1
no change /opt/software/Miniconda3/4.9.2/shell/condabin/conda-hook.ps1
no change /opt/software/Miniconda3/4.9.2/lib/python3.8/site-packages/xontrib/conda.xsh
no change /opt/software/Miniconda3/4.9.2/etc/profile.d/conda.csh
modified /home/jillashey/.bashrc
==> For changes to take effect, close and re-open your current shell. <==
I closed the terminal window and logged back in on a new window. Now let’s activate!
conda activate /data/putnamlab/miranda
Now the conda environment is activated! My shell thing now looks like this: (/data/putnamlab/miranda) [jillashey@ssh3 putnamlab]$. To deactivate, do conda deactivate.
Create a conda environment for mirdeep2 using the same steps above.
I’m going to first work in the mirdeep2 environment. Activate the environment: conda activate /data/putnamlab/mirdeep2
Install mirdeep2 within the conda env: conda install bioconda::mirdeep2. This will take a few minutes to install and load the required packages.
I’m going to try to run mirDeep2 using code from the mirdeep2 github tutorial and Sam White’s code from the E5 deep dive project.
Before running any mirdeep2 modules, I need to upload some databases to the HPC and configure some of my files. Let’s first configure my files. There are 2 files (R1 and R2) per sample, so I need to concatenate and collapse the reads.
cat trimmed.AST-1065_R1_001.fastq.gz trimmed.AST-1065_R2_001.fastq.gz > cat.trimmed.AST-1065.fastq
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
# gunzip cat.trimmed.AST-1065.fastq.gz # files must be unzipped for collapsing; unzip if needed
fastx_collapser -v -i cat.trimmed.AST-1065.fastq -o collapse.cat.trimmed.AST-1065.fastq
head collapse.cat.trimmed.AST-1065.fastq
>1-116635
TGGTCTATGGTGTAACTGGCAACACGTCTGT
>2-115039
ACAGACGTGTTGCCAGTTACACCATAGACCA
>3-104350
TGGTCTATGGTGTAACTGGCAACACGTCTGTT
>4-103158
AACAGACGTGTTGCCAGTTACACCATAGACCA
>5-71882
TGAAAATCTTTTCTCTGAAGTGGAA
As per the mirdeep2 documentation, The readID must end with _xNumber and is not allowed to contain whitespaces. So it has to have the format name_uniqueNumber_xnumber.
sed '/^>/ s/-/_x/g' collapse.cat.trimmed.AST-1065.fastq \
| sed '/^>/ s/>/>seq_/' \
> collapse.cat.trimmed.AST-1065.fastq
>seq_1_x116635
TGGTCTATGGTGTAACTGGCAACACGTCTGT
>seq_2_x115039
ACAGACGTGTTGCCAGTTACACCATAGACCA
>seq_3_x104350
TGGTCTATGGTGTAACTGGCAACACGTCTGTT
>seq_4_x103158
AACAGACGTGTTGCCAGTTACACCATAGACCA
>seq_5_x71882
TGAAAATCTTTTCTCTGAAGTGGAA
Next I need to reformat the genome fasta description lines. miRDeep2 can’t process genome FastAs with spaces in the description lines. I don’t think the Apoc genome has any spaces but I’m going to double check.
cd /data/putnamlab/jillashey/Astrangia_Genome/
grep "^>" apoculata.assembly.scaffolds_chromosome_level.fasta
>chromosome_1
>chromosome_2
>chromosome_3
>chromosome_4
>chromosome_5
>chromosome_6
>chromosome_7
>chromosome_8
>chromosome_9
>chromosome_10
>chromosome_11
>chromosome_12
>chromosome_13
>chromosome_14
Nice, there are no spaces so I don’t need to reformat. If I did, I would sub the spaces with underscores.
Index the genome with bowtie (NOT bowtie2). In the scripts folder: nano bowtie_build.sh
#!/bin/bash
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.out
module load GCCcore/11.3.0 #I needed to add this to resolve conflicts between loaded GCCcore/9.3.0 and GCCcore/11.3.0
module load Bowtie/1.3.1-GCC-11.3.0
# Index the reference genome for A. poculata
bowtie-build /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta Apoc_ref.btindex
echo "Referece genome indexed!" $(date)
The indexed genome lives in the scripts folder for now.
Load the miRbase mature miRNA fasta database onto the server. I downloaded it onto my computer (its not very large) and will now copy it to the server. I downloaded it on 1/3/24. It will live in the refs folder in the smRNA directory.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA
mkdir refs
cd refs
ls refs
20240103_mature.fa
head 20240103_mature.fa
>cel-let-7-5p MIMAT0000001 Caenorhabditis elegans let-7-5p
UGAGGUAGUAGGUUGUAUAGUU
>cel-let-7-3p MIMAT0015091 Caenorhabditis elegans let-7-3p
CUAUGCAAUUUUCUACCUUACC
>cel-lin-4-5p MIMAT0000002 Caenorhabditis elegans lin-4-5p
UCCCUGAGACCUCAAGUGUGA
>cel-lin-4-3p MIMAT0015092 Caenorhabditis elegans lin-4-3p
ACACCUGGGCUCUCCGGGUACC
>cel-miR-1-5p MIMAT0020301 Caenorhabditis elegans miR-1-5p
CAUACUUCCUUACAUGCCCAUA
Check how many mature miRNA sequences there are in the file
zgrep -c ">" 20240103_mature.fa
48885
I’m going to reformat the fasta header names here so there are no spaces
sed '/^>/ s/ /_/g' 20240103_mature.fa \
| sed '/^>/ s/,//g' \
> 20240103_mature.fa
head 20240103_mature.fa
>cel-let-7-5p_MIMAT0000001_Caenorhabditis_elegans_let-7-5p
UGAGGUAGUAGGUUGUAUAGUU
>cel-let-7-3p_MIMAT0015091_Caenorhabditis_elegans_let-7-3p
CUAUGCAAUUUUCUACCUUACC
>cel-lin-4-5p_MIMAT0000002_Caenorhabditis_elegans_lin-4-5p
UCCCUGAGACCUCAAGUGUGA
>cel-lin-4-3p_MIMAT0015092_Caenorhabditis_elegans_lin-4-3p
ACACCUGGGCUCUCCGGGUACC
>cel-miR-1-5p_MIMAT0020301_Caenorhabditis_elegans_miR-1-5p
CAUACUUCCUUACAUGCCCAUA
Do I need to change the U to T in 20240103_mature.fa? Let’s try mapping first and seeing.
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/collapse.cat.trimmed.AST-1065.fastq -e -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s reads_collapsed.fa -t reads_collapsed_vs_genome.arf -v
Didn’t take very long! Only a few seconds. It output this:
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_10292
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 4289826 379347 3910479 8.843 91.157
seq: 4289826 379347 3910479 8.843 91.157
Not a very high mapping rate but I wonder if that’s normal. I may need to change the U to T in the miRbase file. It also produced a bowtie.log file:
less bowtie.log
# reads processed: 1768
# reads with at least one reported alignment: 215 (12.16%)
# reads that failed to align: 1496 (84.62%)
# reads with alignments suppressed due to -m: 57 (3.22%)
Reported 613 alignments to 1 output stream(s)
The reads_collapsed_vs_genome.arf provides info about the sequences that did align:
head reads_collapsed_vs_genome.arf
seq_19_x21706 32 1 32 aacttttgacggtggatctcttggctcacgca chromosome_2 32 42321 42352 aacttttgacggtggatctcttggctcacgca - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x21706 32 1 32 aacttttgacggtggatctcttggctcacgca chromosome_2 32 53070 53101 aacttttgacggtggatctcttggctcacgca - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x21706 32 1 32 aacttttgacggtggatctcttggctcacgca chromosome_2 32 20734 20765 aacttttgacggtggatctcttggctcacgca - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x21706 32 1 32 aacttttgacggtggatctcttggctcacgca chromosome_2 32 31478 31509 aacttttgacggtggatctcttggctcacgca - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x21513 32 1 32 tgcgtgagccaagagatccaccgtcaaaagtt chromosome_2 32 31478 31509 tgcgtgagccaagagatccaccgtcaaaagtt + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x21513 32 1 32 tgcgtgagccaagagatccaccgtcaaaagtt chromosome_2 32 20734 20765 tgcgtgagccaagagatccaccgtcaaaagtt + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x21513 32 1 32 tgcgtgagccaagagatccaccgtcaaaagtt chromosome_2 32 42321 42352 tgcgtgagccaagagatccaccgtcaaaagtt + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x21513 32 1 32 tgcgtgagccaagagatccaccgtcaaaagtt chromosome_2 32 53070 53101 tgcgtgagccaagagatccaccgtcaaaagtt + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_33_x17087 43 1 43 ttgctacgatcttctgagattaagcctttgttctaagatttgt chromosome_2 43 879093 879135 ttgctacgatcttctgagattaagcctttgttctaagatttgt +mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_33_x17087 43 1 43 ttgctacgatcttctgagattaagcctttgttctaagatttgt chromosome_2 43 38360 38402 ttgctacgatcttctgagattaagcctttgttctaagatttgt -mmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
Not sure what all of this info means, but I will look into it. I also need to look into why/how the reads get collapsed because the collapse set left me with only 1796 sequences (compared to the 26561348 sequences I had in the cat file) and I want to make sure that’s normal.
Now lets run mirdeep2!!!!!!!!
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/collapse.cat.trimmed.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature.fa none none -t N.vectensis -P -v -g -1 2>report.log
I need to specify none 2x because I do not have the files for known miRNAs or known precursor miRNAs in this species. I got an error:
#Starting miRDeep2
/data/putnamlab/mirdeep2/bin/miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/collapse.cat.trimmed.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature.fa none none -t N.vectensis -P -v -g -1
miRDeep2 started at 16:15:57
mkdir mirdeep_runs/run_04_01_2024_t_16_15_57
#testing input files
started: 16:16:06
sanity_check_mature_ref.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature.fa
ended: 16:16:06
total:0h:0m:0s
sanity_check_reads_ready_file.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/collapse.cat.trimmed.AST-1065.fastq
started: 16:16:06
ESC[1;31mError: ESC[0mproblem with /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/collapse.cat.trimmed.AST-1065.fastq
Error in line 860: Either the sequence
GATGGAATTGTAGCAT
contains less than 17 characters or contains characters others than [acgtunACGTUN]
Please make sure that your file only comprises sequences that have at least 17 characters
containing letters [acgtunACGTUN]
My collapsed read file has some sequences that are <17 bp which mirdeep2 doesn’t like. I need to remove sequences with <17 nts (or do it during the trimming step). Used chatgpt for the code below :)
#!/bin/bash
# Define the input and output files
input_file="collapse.cat.trimmed.AST-1065.fastq"
output_file="17_collapse.cat.trimmed.AST-1065.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
Rerun mirdeep2 with the new file
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/17_collapse.cat.trimmed.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature.fa none none -t N.vectensis -P -v -g -1 2>report.log
Took about 2 mins and there were no alignments. I’m going to change the U to T in the 20240103_mature.fa file, then rerun the mapping and mirdeep2 steps.
#!/bin/bash
# Define the input and output files
input_file="20240103_mature.fa" # Replace with your actual input file name
output_file="20240103_mature_T.fa"
# Initialize the output file
> "$output_file"
# Use awk to process the file
awk '{
if (substr($0, 1, 1) == ">") {
print $0 >> "'$output_file'" # Print the identifier as is
} else {
gsub(/U/, "T", $0) # Replace U with T in sequences
print $0 >> "'$output_file'"
}
}' "$input_file"
I guess I don’t need to redo the mapping step because the mapping did not use the 20240103_mature.fa file. Therefore, I will proceed to the mirdeep2 step.
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/fastp/17_collapse.cat.trimmed.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
Still no alignment :’( I need to talk to Sam and ask him the following:
- How many reads were left after he concatnated and collapsed his fastq files?
- What was his alignment after the mapping step?
Also what if I didn’t collapse the reads? What if I just removed the heading, + sign, and quality scores and formatted it like mirdeep2 wants it?
I just briefly reran the collapse step and now there are hundreds of thousands of sequences…may need to rerun mapping idk
20240105
Flexbar finished running overnight, took about 9 hours. Now I need to QC it. First going to move it from the raw data folder to the trim data folder.
In /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim, I moved the old flexbar trimmed seqs to the foler flexbar_old. I moved the newly trimmed seqs from the raw data folder into /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar.
Now let’s run fastqc on the newly trimmed samples using the fastqc_trim.sh from above, but changing the directories
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.out
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
echo "QC for trimmed reads using flexbar with max length of 30 bp" $(date)
for file in /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Astrangia2021/smRNA/fastqc/trim/flexbar
done
echo "QC complete, run multiqc" $(date)
multiqc --interactive fastqc_results/trim/flexbar
Submitted batch job 292159
If the data looks good, I will do another test run of mirdeep2. It finished in ~30 mins but it did not complete the multiQC. When I went into the folder to run the multiqc step, it appears to only have run it on the R2 files. I’m going to move the R1 and R2 fastqc info into separate folders and run the QC on them separately. There is probably a better way to do this idk.
mkdir R1 R2
mv *_1_fastqc* R1
mv *_2_fastqc* R2
Now go into each folder and run multiqc separately. QC looks good for both reads.
Next I need to cat, collapse and prep reads for mirdeep2.
20240107
I’m going to write a script that will cat and collapse the reads. I’ll do it on a test sample first. In the scripts folder: nano test_cat_collapse.sh.
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
echo "Concatenating R1 and R2 for test sample" $(date)
cat AST-1065_R1_001.fastq.gz_1.fastq AST-1065_R1_001.fastq.gz_2.fastq > cat.AST-1065.fastq
echo "Collapsing redundant sequences with fastx collapse" $(date)
fastx_collapser -v -i cat.AST-1065.fastq -o collapse.cat.AST-1065.fastq
echo "Prep sequence IDs for mirdeep2 analysis" $(date)
sed '/^>/ s/-/_x/g' collapse.cat.AST-1065.fastq \
| sed '/^>/ s/>/>seq_/' \
> collapse.cat.AST-1065.fastq
echo "Done!" $(date)
Submitted batch job 292222. I did the genome and database prep already so I don’t need to redo that. Took about 20 mins, but the collapsed file is empty…going to have the output file for the sed portion be sed.collapse.cat.AST-1065.fastq to see if the sed portion is what is happening to the files. Submitted batch job 292224. That iteration worked!
Check how many sequences are in the collapsed file.
zgrep -c ">" sed.collapse.cat.AST-1065.fastq
11979585
head sed.collapse.cat.AST-1065.fastq
>seq_1_x357414
TGGTCTATGGTGTAACTGGCAACACGTCTG
>seq_2_x138955
ACAGACGTGTTGCCAGTTACACCATAGACC
>seq_3_x125294
AACAGACGTGTTGCCAGTTACACCATAGAC
>seq_4_x98253
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_5_x87633
TTCCACTTCAGAGAAAAGATTTTCA
Nice, almost 12 million sequences were retained. I was looking at the fastx collapse documentation and it said that the first number in the sequence id corresponded to a sequence and the second number corresponded to how many times that sequence appeared prior to the file being collapsed. So for example, >seq_1_x357414 was the most represented sequence, as indicated by the 1, and it appeared 357414 times in the pre-collapsed file.
Need to make sure that all of my sequences are >17 bp, as mirdeep2 does not run if sequences are present with <16 bp.
My collapsed read file has some sequences that are <17 bp which mirdeep2 doesn’t like. I need to remove sequences with <17 nts (or do it during the trimming step). Used chatgpt for the code below :)
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1065.fastq"
output_file="17_sed.collapse.cat.AST-1065.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1065.fastq
11979585
Retained all of the sequences. NOW lets attempt an mirdeep2 run.
conda activate /data/putnamlab/mirdeep2
Map reads to genome first
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s 20240107_reads_collapsed.fa -t 20240107_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_56091
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 35658222 3275049 32383173 9.185 90.815
seq: 35658222 3275049 32383173 9.185 90.815
Still got a pretty high % of unmapped reads…lets look at some of the files produced.
head 20240107_reads_collapsed.fa
>seq_1_x357414
TGGTCTATGGTGTAACTGGCAACACGTCTG
>seq_2_x138955
ACAGACGTGTTGCCAGTTACACCATAGACC
>seq_3_x125294
AACAGACGTGTTGCCAGTTACACCATAGAC
>seq_4_x98253
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_5_x87633
TTCCACTTCAGAGAAAAGATTTTCA
zgrep -c ">" 20240107_reads_collapsed.fa
11979585
head 20240107_reads_collapsed_vs_genome.arf
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 27510 27539 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 49105 49134 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 38360 38389 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 879106 879135 acaaatcttagaacaaaggcttaatctcag - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_25_x32759 30 1 30 ttgctacgatcttctgagattaagcctttg chromosome_2 30 879093 879122 ttgctacgatcttctgagattaagcctttg + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_25_x32759 30 1 30 ttgctacgatcttctgagattaagcctttg chromosome_2 30 38373 38402 ttgctacgatcttctgagattaagcctttg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
wc -l 20240107_reads_collapsed_vs_genome.arf
1091161 20240107_reads_collapsed_vs_genome.arf
I’m not sure what the 20240107_reads_collapsed_vs_genome.arf file means. Let’s see how many unique sequences are in that file
cut -f1 20240107_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
523747
So ~500,000 unique sequences were mapped to the genome? That is how I am interpreting this. Let’s try to run mirdeep2.
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/20240107_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
I’m going to let it run for ~10 mins then cut it off, as it probably takes a while (Sam White said his script took several days). I’m not sure if I can activate a conda env in a job script, emailed Kevin Bryan to ask.
Cut the script off, this is as far as it got:
#####################################
# #
# miRDeep2.0.1.3 #
# #
# last change: 08/11/2019 #
# #
#####################################
miRDeep2 started at 17:59:09
#Starting miRDeep2
#testing input files
#parsing genome mappings
#excising precursors
#preparing signature
#folding precursors
Yay, things are happening! Hopefully I can run this by the e5 meeting on Friday.
20240107
Kevin Bryan confirmed that I can run a conda environment in a job script, I just need to add -i to the #!/bin/bash because of the way conda changes environments. Going to try this now on the test smRNA sample. In the scripts folder: nano test_mirdeep2.sh.
#!/bin/bash -i
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on test sample" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/20240107_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for test sample" $(date)
conda deactivate
Submitted batch job 292242. Job has been pending for a few mins and says its waiting for resources.
20230109
After pending for about a day, mirdeep2 finally finished running on the test sample. It took about 3.5 hours to run. It created these folders/files in the scripts folder:
drwxr-xr-x. 3 jillashey 4.0K Jan 9 07:49 mirdeep_runs
drwxr-xr-x. 2 jillashey 4.0K Jan 9 07:52 dir_prepare_signature1704804676
-rw-r--r--. 1 jillashey 377 Jan 9 11:23 error_09_01_2024_t_07_49_09.log
-rw-r--r--. 1 jillashey 63K Jan 9 11:24 result_09_01_2024_t_07_49_09.csv
-rw-r--r--. 1 jillashey 704K Jan 9 11:24 result_09_01_2024_t_07_49_09.html
drwxr-xr-x. 2 jillashey 4.0K Jan 9 11:28 pdfs_09_01_2024_t_07_49_09
-rw-r--r--. 1 jillashey 28K Jan 9 11:28 result_09_01_2024_t_07_49_09.bed
drwxr-xr-x. 2 jillashey 4.0K Jan 9 11:28 mirna_results_09_01_2024_t_07_49_09
-rw-r--r--. 1 jillashey 20K Jan 9 11:28 report.log
Let’s look at them each.
cd mirdeep_runs
cd run_09_01_2024_t_07_49_09 # folder
ls
identified_precursors.fa output.mrd rfam_vs_precursor.bwt run_09_01_2024_t_07_49_09_parameters survey.csv
The identified precursors fasta file includes the precursor sequences (ie the part of the sequence that forms the pre-miRNA)
head identified_precursors.fa
>chromosome_10_48090
ugauggagauggagaacgagaguggacuggacaguuuggcacugaagguucccuuuauaagcaguguuuuucuuucgacuacc
>M:chromosome_10_48090
TGTTTTTCTTTCGACTACC
>L:chromosome_10_48090
AGTGGACTGGACAGTTTGGCACTGAAGGTTCCCTTTATAAGCAG
>S:chromosome_10_48090
TGATGGAGATGGAGAACGAG
>chromosome_14_108653
cgcgcgcuauaguuacaguagcuauagcgcgcacuauaauuauagcagcuauagcgcacgcuauaguuagaaacuguagcgcgaguu
zgrep -c ">" identified_precursors.fa
18695
Almost 19000 precursor sequences.
In the output.mrd file, it has info on the different miRNAs identified I believe
>chromosome_7_30929
score total 2.4
score for star read(s) -1.3
score for read counts 0
score for mfe 2.1
score for randfold 1.6
total read count 13651
mature read count 13499
loop read count 0
star read count 152
exp fffffffffffffffffffMMMMMMMMMMMMMMMMMMMMlllllllllllllllSSSSSSSSSSSSSSSSSSSSffffffffffffffffffffffffff
ffffffffffff
obs fffffffffffffffffffMMMMMMMMMMMMMMMMMMMMlllllllllllllSSSSSSSSSSSSSSSSSSSSSfffffffffffffffffffffffffff
ffffffffffff
pri_seq cgcacugcaguugacgugaacccguagauccgaacuugugggauuuuucuccacaaguucggcuccaugguccacgugugcugugcucacaaacguugcu
acagcgugguca
pri_struct .((((.(((....(((((...((((.((.((((((((((((((....)).)))))))))))).)).))))..))))).))).))))......((((((..
.))))))..... #MM
seq_7183221_x1 .................Uaacccguagauccgaacuugug............................................................
............ 1
seq_2180915_x2 ..................aacccguagauccgaacu................................................................
............ 0
seq_9719163_x1 ..................aacccguagauccgaUcuu...............................................................
............ 1
seq_2267835_x2 ..................Cacccguagauccgaacuu...............................................................
............ 1
ola-miR-100_MIMAT0022614_Oryzias_latipes_miR-100 ..................aacccguagauccgaacuu...............................................................
............ 0
seq_126867_x21 ..................aacccguagauccgaacuug..............................................................
............ 0
seq_254962_x11 ..................Cacccguagauccgaacuug..............................................................
............ 1
sbo-miR-100_MIMAT0049501_Saimiri_boliviensis_miR-100 ..................aacccguagauccgaacuugu.............................................................
............ 0
dma-miR-100_MIMAT0049252_Daubentonia_madagascariensis_miR-100 ..................aacccguagauccgaacuugu.............................................................
............ 0
seq_199802_x14 ..................aacccguagauccgaacuugC.............................................................
............ 1
pmi-miR-100-5p_MIMAT0032156_Patiria_miniata_miR-100-5p ..................aacccguagauccgaacuugu.............................................................
............ 0
seq_2153292_x2 ..................aacccguagauccgaGcuugu.............................................................
............ 1
seq_7747127_x1
zgrep -c ">" output.mrd
5231
The rfam vs precursor file includes information about where on the chromosomes the rRNAs and tRNAs are?
head rfam_vs_precursor.bwt
M:chromosome_13_66113 + AM086652.1/1-576_RF00177;SSU_rRNA_5; 468 TGTTTCGGGATTGCAATG IIIIIIIIIIIIIIIIII 3
M:chromosome_13_66113 + AF508778.1/21-597_RF00177;SSU_rRNA_5; 469 TGTTTCGGGATTGCAATG IIIIIIIIIIIIIIIIII 3
M:chromosome_13_66113 + AJ310485.1/21-596_RF00177;SSU_rRNA_5; 468 TGTTTCGGGATTGCAATG IIIIIIIIIIIIIIIIII 3
M:chromosome_13_66113 + DQ057346.1/21-597_RF00177;SSU_rRNA_5; 469 TGTTTCGGGATTGCAATG IIIIIIIIIIIIIIIIII 3
S:chromosome_13_66113 + AACY021626480.1/155-83_RF00005;tRNA; 26 TTTGTTTCGTAAGCAAA IIIIIIIIIIIIIIIII 2 7:T>C
S:chromosome_13_66113 + AACY023301721.1/825-896_RF00005;tRNA; 25 TTTGTTTCGTAAGCAAA IIIIIIIIIIIIIIIII 2 12:A>G
S:chromosome_13_66113 + AACY022901721.1/116-188_RF00005;tRNA; 26 TTTGTTTCGTAAGCAAA IIIIIIIIIIIIIIIII 2 12:A>G
M:chromosome_14_72583 + CP000030.1/153914-153986_RF00005;tRNA; 3 CGGTTAGCTCAGTTGGTAGA IIIIIIIIIIIIIIIIIIII 13
M:chromosome_14_72583 + AACY020037993.1/1235-1163_RF00005;tRNA; 3 CGGTTAGCTCAGTTGGTAGA IIIIIIIIIIIIIIIIIIII 13
M:chromosome_14_72583 + AACY020166163.1/13-85_RF00005;tRNA; 3 CGGTTAGCTCAGTTGGTAGA IIIIIIIIIIIIIIIIIIII 13
wc -l rfam_vs_precursor.bwt
2241 rfam_vs_precursor.bwt
The run parameters file has the code specifics
Start: 09_01_2024_t_07_49_09
Script /data/putnamlab/mirdeep2/bin/miRDeep2.pl
args /data/putnamlab/mirdeep2/bin/miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/20240107_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature_T.fa none none -t N.vectensis -P -v -g -1
dir_with_tmp_files dir_miRDeep2_09_01_2024_t_07_49_09
dir /glfs/brick01/gv0/putnamlab/jillashey/Astrangia2021/smRNA/scripts
file_reads /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq
file_genome /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta
file_reads_vs_genome /data/putnamlab/jillashey/Astrangia2021/smRNA/20240107_reads_collapsed_vs_genome.arf
file_mature_ref_this_species /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/20240103_mature_T.fa
file_mature_ref_other_species none
option{t} = N.vectensis
option{v} = used
miRDeep runtime:
started: 7:49:09
ended: 11:28:44
total:3h:39m:35s
The survey file inclues info about the mirdeep2 scores. This is the same info that is at the top of the hmtl and csv files.
miRDeep2 score novel miRNAs reported by miRDeep2 novel miRNAs, estimated false positives novel miRNAs, estimated true positives known miRNAs in species known miRNAs in data known miRNAs detected by miRDeep2 estimated signal-to-noise excision gearing
10 49 3 +/- 2 46 +/- 2 (93 +/- 3%) 48885 83 1 (1%) 15.6 1
9 51 3 +/- 2 48 +/- 2 (93 +/- 3%) 48885 83 1 (1%) 15.1 1
8 56 4 +/- 2 52 +/- 2 (93 +/- 3%) 48885 83 1 (1%) 15.4 1
7 64 4 +/- 2 60 +/- 2 (94 +/- 3%) 48885 83 1 (1%) 16.3 1
6 68 4 +/- 2 64 +/- 2 (94 +/- 3%) 48885 83 1 (1%) 16 1
5 70 5 +/- 2 65 +/- 2 (93 +/- 3%) 48885 83 1 (1%) 15 1
4 72 5 +/- 2 67 +/- 2 (93 +/- 3%) 48885 83 1 (1%) 14 1
3 101 6 +/- 2 95 +/- 2 (94 +/- 2%) 48885 83 1 (1%) 16.2 1
2 183 9 +/- 3 174 +/- 3 (95 +/- 2%) 48885 83 72 (87%) 20.2 1
1 260 22 +/- 4 238 +/- 4 (91 +/- 2%) 48885 83 72 (87%) 11.6 1
0 315 53 +/- 6 262 +/- 6 (83 +/- 2%) 48885 83 72 (87%) 5.9 1
-1 365 97 +/- 8 268 +/- 8 (73 +/- 2%) 48885 83 72 (87%) 3.8 1
-2 462 150 +/- 10 312 +/- 10 (67 +/- 2%) 48885 83 72 (87%) 3.1 1
-3 707 228 +/- 14 479 +/- 14 (68 +/- 2%) 48885 83 72 (87%) 3.1 1
-4 1003 402 +/- 18 601 +/- 18 (60 +/- 2%) 48885 83 73 (88%) 2.5 1
-5 1167 750 +/- 23 417 +/- 23 (36 +/- 2%) 48885 83 73 (88%) 1.6 1
-6 1309 1227 +/- 35 82 +/- 35 (6 +/- 3%) 48885 83 73 (88%) 1.1 1
-7 1405 1733 +/- 42 0 +/- 0 (0 +/- 0%) 48885 83 73 (88%) 0.8 1
-8 1653 2208 +/- 45 0 +/- 0 (0 +/- 0%) 48885 83 73 (88%) 0.7 1
-9 2013 2615 +/- 49 0 +/- 0 (0 +/- 0%) 48885 83 73 (88%) 0.8 1
-10 2423 2951 +/- 53 0 +/- 0 (0 +/- 0%) 48885 83 73 (88%) 0.8 1
Going into the dir_prepare_signature1704804676 from the scripts folder
cd dir_prepare_signature1704804676
ls
mature_vs_precursors.arf precursors.ebwt.2.ebwt precursors.ebwt.rev.1.ebwt reads_vs_precursors.arf signature_unsorted.arf.tmp
mature_vs_precursors.bwt precursors.ebwt.3.ebwt precursors.ebwt.rev.2.ebwt reads_vs_precursors.bwt signature_unsorted.arf.tmp2
precursors.ebwt.1.ebwt precursors.ebwt.4.ebwt precursors.fa signature_unsorted.arf
Looked at the mature vs. precursors file
head mature_vs_precursors.arf
hsa-miR-100-5p_MIMAT0000098_Homo_sapiens_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30929 22 19 40 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
hsa-miR-100-5p_MIMAT0000098_Homo_sapiens_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30930 22 69 90 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
mmu-miR-100-5p_MIMAT0000655_Mus_musculus_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30930 22 69 90 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
mmu-miR-100-5p_MIMAT0000655_Mus_musculus_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30929 22 19 40 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
rno-miR-100-5p_MIMAT0000822_Rattus_norvegicus_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30930 22 69 90 aacccgtagatccgaacttgtg + 0 mmmmmmmmmmmmmmmmmmmmmm
rno-miR-100-5p_MIMAT0000822_Rattus_norvegicus_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30929 22 19 40 aacccgtagatccgaacttgtg + 0 mmmmmmmmmmmmmmmmmmmmmm
gga-miR-100-5p_MIMAT0001178_Gallus_gallus_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30930 22 69 90 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
gga-miR-100-5p_MIMAT0001178_Gallus_gallus_miR-100-5p 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30929 22 19 40 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
aga-miR-100_MIMAT0001498_Anopheles_gambiae_miR-100 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30930 22 69 90 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
aga-miR-100_MIMAT0001498_Anopheles_gambiae_miR-100 22 1 22 aacccgtagatccgaacttgtg chromosome_7_30929 22 19 40 aacccgtagatccgaacttgtg +mmmmmmmmmmmmmmmmmmmmmm
wc -l mature_vs_precursors.arf
191 mature_vs_precursors.arf
I’m not sure what this means…Need to look into this more. Is it providing info about the mirbase sequences in comparison to my own? It looks like most of them are related to miR-100, which makes sense as this is the only miRNA that is in bilaterians and cnidarians.
Back in the scripts directory, look at the error file:
RNAfold: invalid option -- n
total number of rounds controls=100
1^M2^M3^M4^M5^M6^M7^M8^M9^M10^M11^M12^M13^M14^M15^M16^M17^M18^M19^M20^M21^M22^M23^M24^M25^M26^M27^M28^M29^M30^M31^M32^M33^M34^M35^M36^M37^M38^M39^M40^M41^M42^M43^M44^M45^M46^M47^M48^M49^M50^M51^M52^M53^M54^M55^M56^M57^M58^M59^M60^M61^M62^M63^M64^M65^M66^M67^M68^M69^M70^M71^M72^M73^M74^M75^M76^M77^M78^M79^M80^M81^M82^M83^M84^M85^M86^M87^M88^M89^M90^M91
^M92^M93^M94^M95^M96^M97^M98^M99^M100^Mcontrols performed
Not sure what this means either…some issue with the RNAfold option? But I got a randfold pvalue.
The pdf folder includes a pdf file for each miRNA (?) identified and provides info about the scores and gives a nice graph about where the mature and star sequences are
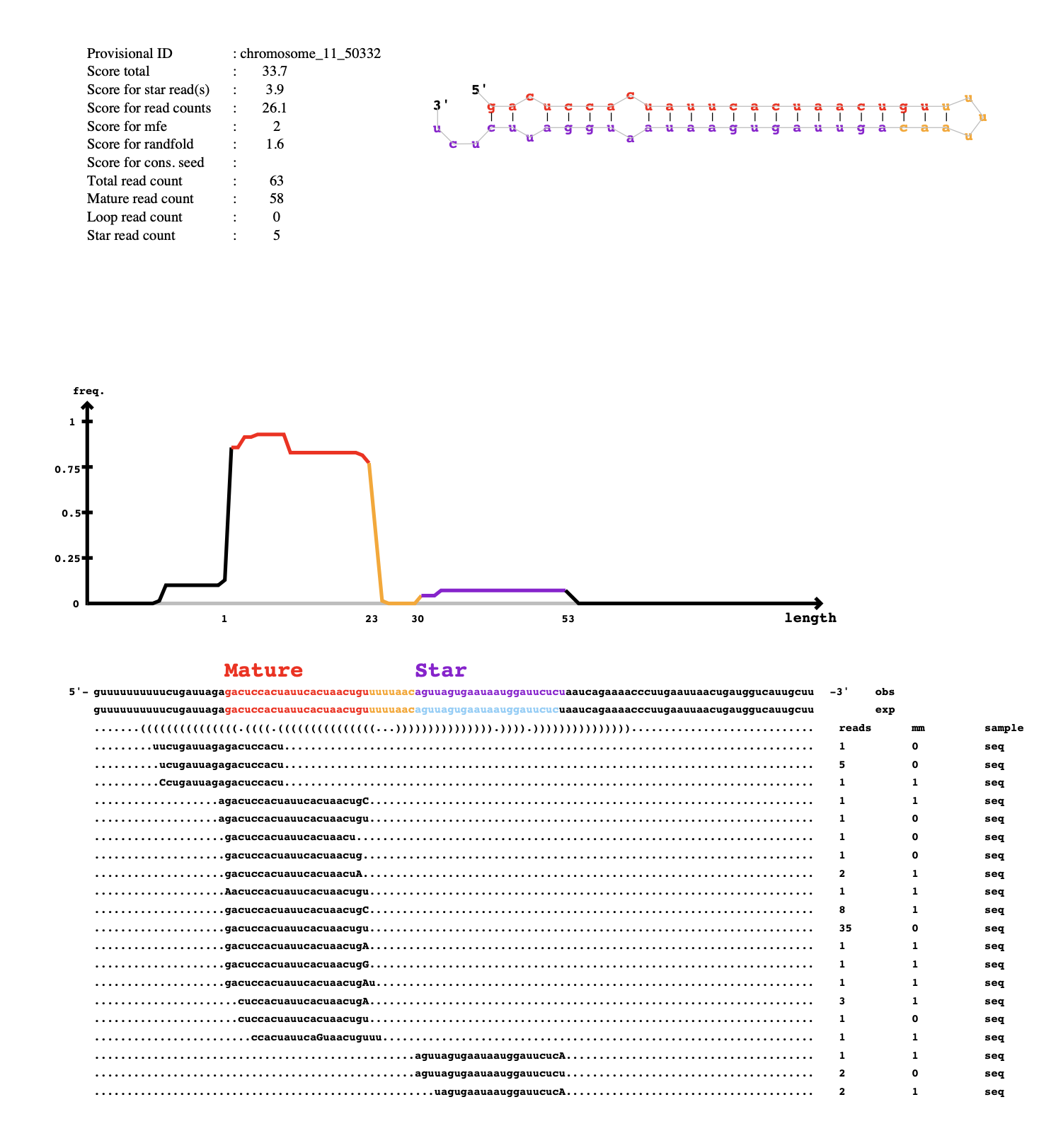
In my first run of mirdeep2, 318 unique pdfs were produced. In the mirna_results_09_01_2024_t_07_49_09 results folder, there are .bed and .fa files. The bed files contain this info:
less known_mature_09_01_2024_t_07_49_09_score-50_to_na.bed
browser position chromosome_6:21902755-21902777
browser hide all
track name="notTrackname.known_miRNAs" description="known miRNAs detected by miRDeep2 for notTrackname" visibility=2
itemRgb="On";
chromosome_6 21902755 21902777 chromosome_6_22810 19.4 + 21902755 21902777 255,0,0
chromosome_7 21599230 21599250 chromosome_7_30929 2.4 - 21599230 21599250 0,0,255
chromosome_11 24411396 24411418 chromosome_11_54894 -3.5 - 24411396 24411418 0,0,255
and the fa files contain this info:
less known_mature_09_01_2024_t_07_49_09_score-50_to_na.fa
>chromosome_6_22810
AAGAACACCCAAAATAGCTGAA
>chromosome_7_30929
ACCCGTAGATCCGAACTTGT
>chromosome_11_54894
GCGGGTGTGTGTGTGTGTGTGT
So I suppose its giving information about the known mature sequences and their location on the Astrangia genome. The other files in this folder contain bed and fa files for the known precursors, star and mature sequences, as well as the novel precursors, star and mature sequences.
In the main scripts folder, there is also a file result_09_01_2024_t_07_49_09 in .bed, .csv and .html format. It has the same info in all of them, but it is summarizing the parameters used, the survey info, the novel miRNAs and the known miRNAs. In the survey info, it says that 83 known miRNAs were identified in my data but only 3 are listed at the bottom?
Anything that is listed as mirdeep2 results from Jan 9 are associated with AST-1065.
I wonder if these results would change if I trimmed to 25 bp. I also wonder what would happen if I removed Nvectensis as my related species. Sam white put S.purpurtus as the related species, but Nvectensis is more closely related. I’m going to remove the related species and rerun mirdeep2. Submitted batch job 292316. Started running immediately, nice.
I’m also going to rerun flexbar with trimming of 25 bp. In the trim data folder, make a folder for flexbar 25 bp
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim
mkdir flexbar_25bp
In the scripts folder: nano flexbar_25bp.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=200GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Flexbar/3.5.0-foss-2018b
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
echo "Trimming reads to 25 bp using flexbar" $(date)
array1=($(ls *R1_001.fastq.gz))
for i in ${array1[@]}; do
flexbar \
-r ${i} \
-p $(echo ${i}|sed s/_R1/_R2/) \
-a NEB-adapters.fasta \
-ap ON \
-qf i1.8 \
-qt 30 \
-t /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar_25bp \
-k 25 \
-R trim.${i} \
-P trim.$(echo ${i}|sed s/_R1/_R2/) \
-z GZ
done
echo "Trimming complete" $(date)
Submitted batch job 292317. Immediately got an error saying: ERROR: Could not open file trim.AST-2360_R1_001.fastq.gz.gz. I commented out the -z command. Submitted batch job 292344
20230110
Hooray! Flexbar 25 bp and mirdeep2 test finished running. Let’s look at the mirdeep2 results. As a reminder, I reran the mirdeep2 code but removed the specification of Nematostella as a related species. When removing this specification, I got marginally more (1-2 more) novel miRNAs predicted, but got the same number of known miRNAs identified. The mirdeep2 documentation states “it will in practice always improve miRDeep2 performance if miRNAs from some related species is input, even if it is not closely related.” So it is likely best to keep Nematostella as a related species in the code.
My next step is to run mirdeep2 on the newly trimmed (25 bp) reads. Again, I’m going to run it on a test sample. First, move the newly trimmed reads from the raw data folder to the flexbar 25 bp folder.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/raw
mv trim* ../trim/flexbar_25bp/
Next run fastqc. In the fastqc folder, make a folder for the new flexbar 25bp QC data.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/fastqc/trim
mkdir flexbar_25bp
cd flexbar_25bp
mkdir R1 R2
In the scripts folder, edit the fastqc_trim.sh script:
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load FastQC/0.11.9-Java-11
module load MultiQC/1.9-intel-2020a-Python-3.8.2
echo "QC for trimmed reads using flexbar with max length of 25 bp" $(date)
for file in /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar_25bp/*fastq.gz
do
fastqc $file --outdir /data/putnamlab/jillashey/Astrangia2021/smRNA/fastqc/trim/flexbar_25bp
done
echo "FastQC complete" $(date)
#multiqc --interactive fastqc_results/trim/flexbar
20240112
Had to rerun the trimming bc I accidently left the 30 as the max length. Now im running the QC. Submitted batch job 292416
Once this has finished running, navigate to the flexbar 25bp fastqc folder and move the reads into R1 and R2 folders.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/fastqc/trim/flexbar_25bp
mv *_R1* R1
mv *_R2* R2
Go into each folder and run MultiQC on each. I have to do this because for some reason, multiQC was just running QC stats on the R2 reads when the R1 and R2 reads were in the same folder.
I think it would be a good idea to make a custom database that includes primarily cnidarian miRNAs, as there are not many in mirbase itself. Baumgarten et al. (2017) did something similar (under 2.3 miRNA annotation in their paper).
- “miRNAs were then annotated using the miRDeep2 package with default settings (Friedlander et al., 2012). To identify putatively conserved miRNAs based on previous de novo annotations of other cnidarian genomes, we created a reference library of mature miRNA sequences from N. vectensis (Grimson et al., 2008; Moran et al., 2014), Hydra magnipapillata (Krishna et al., 2013) and Stylophora pistillata (Liew et al., 2014).”
Therefore, I am going to make a custom database of the cnidarian miRNAs that I am aware of.
- Stylophora pistillata - Table S11 from Liew et al. 2014
- Aiptasia - Table S4 from Baumgarten et al. 2017
- Acropora digitifera - Table S5 from Gajigan & Conaco 2017
- the sea anemones Edwardsiella carnea, Scolanthus callimorphus, Metridium senile and Anemonia viridis, and the stony coral Acropora millepora - Tables S1 and S2 from Praher et al. 2021
- Hydra - Table S1 (SuppFile2) from Krishna et al. 2012
- Nematostella - Table S1 from Moran et al. 2014
- Anemonia viridis - Table S2 from Urbarova et al. 2018
- Nematostella + Hydra are also both on miRBase
In google sheets, I gathered all the cnidarian miRNA sequences that I could find and made it into a csv file. This is the format:
miRNA Mature_miRNA_sequence Species Citation Notes
spi-mir-temp-1 acccguagauccgaacuugugg Stylophora pistillata Liew et al. 2014 Matches miR-100 family.
spi-mir-temp-2 uaucgaauccgucaaaaagaga Stylophora pistillata Liew et al. 2014 NA
spi-mir-temp-3 ucagggauuguggugaguuaguu Stylophora pistillata Liew et al. 2014 NA
spi-mir-temp-4 aaagaaguacaagugguaggg Stylophora pistillata Liew et al. 2014 Exact match of nve-miR-2023.
spi-mir-temp-5 gagguccggaugguuga Stylophora pistillata Liew et al. 2014 NA
I downloaded the csv to my computer and manipulated it so that the first, third, fourth and fifth columns are the headers on one line and denoted with a “>”. Then the sequence, in the second column, was put under the header.
awk -F',' 'NR>1 {print ">"$1" "$2" "$3" "$4"\n"$5}' cnidarian_miRNAs.csv > cnidarian_miRNAs.fasta
Now go back to the miRbase fasta file and subset so that I make a file with Hydra and Nematostella sequences only
awk '/>.*hma|>.*nve/ {print; getline; print}' 20240103_mature_T.fa > subset.fasta
I then copied the subset fasta info into the cnidarian_miRNAs.fasta. A complete cnidarian miRNA fasta! Reformat the fasta header names so there are no spaces.
sed '/^>/ s/ /_/g' cnidarian_miRNAs.fasta \
| sed '/^>/ s/,//g' \
> cnidarian_miRNAs.fasta
Reformat sequences so that everything is uppercase
awk '/^>/ {print; getline; print toupper($0); next} {print}' cnidarian_miRNAs.fasta > cnidarian_miRNAs.fasta
I then copied the file to andromeda to /data/putnamlab/jillashey/Astrangia2021/smRNA/refs. Change the U to T in the fasta file
#!/bin/bash
# Define the input and output files
input_file="cnidarian_miRNA.fa" # Replace with your actual input file name
output_file="cnidarian_miRNA_T.fa"
# Initialize the output file
> "$output_file"
# Use awk to process the file
awk '{
if (substr($0, 1, 1) == ">") {
print $0 >> "'$output_file'" # Print the identifier as is
} else {
gsub(/U/, "T", $0) # Replace U with T in sequences
print $0 >> "'$output_file'"
}
}' "$input_file"
Now I am going to run mirdeep2 with the cnidarian miRNA file! I’m going to modify the nano test_mirdeep2.sh so that the fasta file is the cnidarian_miRNA_T.fa file. Submitted batch job 292356. Didnt work. Removed the -t argument. Submitted batch job 292358…still not running. Getting this error:
bash: cannot set terminal process group (-1): Function not implemented
bash: no job control in this shell
Need to troubleshoot this.
When I look at the report.log file, it says:
miRDeep2 started at 12:39:17
mkdir mirdeep_runs/run_12_01_2024_t_12_39_17
#testing input files
started: 12:39:23
sanity_check_mature_ref.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/cnidarian_miRNA_T.fa
ESC[1;31mError: ESC[0mproblem with /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/cnidarian_miRNA_T.fa
Error in line 64: The sequence
SPIS
contains characters others than [acgtunACGTUN]
Please check your file for the following issues:
I. Sequences are allowed only to comprise characters [ACGTNacgtn].
II. Identifiers are not allowed to have withespaces.
I looked at the cnidarian_miRNA_T.fa file and found that for a few sequences, it has Spis instead of the actual sequence:
>spi-mir-temp-42 Stylophora pistillata Liew et al. 2014 NA
ugugcaagaauuugagucgcugg
>apa-mir-100 Exaiptasia pallida Baumgarten et al. 2017 "miR-100; Nve
Spis
>apa-mir-2022a Exaiptasia pallida Baumgarten et al. 2017 "miR-2022; Nve
Spis
>apa-mir-2023 Exaiptasia pallida Baumgarten et al. 2017 "miR-2023; Nve
Spis
>apa-mir-2025 Exaiptasia pallida Baumgarten et al. 2017 "miR-2025; Nve
Adi"
>apa-mir-2026 Exaiptasia pallida Baumgarten et al. 2017 miR-2026; Nve
aauuucaaauauccacugauug
>apa-mir-2030 Exaiptasia pallida Baumgarten et al. 2017 "miR-2030; Nve
Spis
>apa-mir-2036 Exaiptasia pallida Baumgarten et al. 2017 "miR-2036; Nve
Spis
>apa-mir-2037 Exaiptasia pallida Baumgarten et al. 2017 "miR-2037; Nve
Spis"
>apa-mir-2050 Exaiptasia pallida Baumgarten et al. 2017 "miR-2050; Nve
Spis
I’m thinking maybe it doesn’t like the commas? Going back to csv and adding semi-colans instead of commas.
I downloaded the csv to my computer and manipulated it so that the first, third, fourth and fifth columns are the headers on one line and denoted with a “>”. Then the sequence, in the second column, was put under the header.
awk -F',' 'NR>1 {print ">"$1" "$2" "$3" "$4"\n"$5}' cnidarian_miRNAs.csv > cnidarian_miRNAs.fasta
That seems to have fixed the problem. Reformat the fasta header names so there are no spaces.
sed '/^>/ s/ /_/g' cnidarian_miRNAs.fasta \
| sed '/^>/ s/,//g' \
> cnidarian_miRNAs.fasta
Reformat sequences so that everything is uppercase
awk '/^>/ {print; getline; print toupper($0); next} {print}' cnidarian_miRNAs.fasta > cnidarian_miRNAs.fasta
I then copied the file to andromeda to /data/putnamlab/jillashey/Astrangia2021/smRNA/refs. Change the U to T in the fasta file
#!/bin/bash
# Define the input and output files
input_file="cnidarian_miRNAs.fa" # Replace with your actual input file name
output_file="cnidarian_miRNAs_T.fa"
# Initialize the output file
> "$output_file"
# Use awk to process the file
awk '{
if (substr($0, 1, 1) == ">") {
print $0 >> "'$output_file'" # Print the identifier as is
} else {
gsub(/U/, "T", $0) # Replace U with T in sequences
print $0 >> "'$output_file'"
}
}' "$input_file"
Now concatenate the cnidarian miRNAs with the mature miRNA fasta from miRBase.
cat 20240103_mature_T.fa cnidarian_miRNAs_T.fa > mature_mirbase_cnidarian_T.fa
Now edit the test_mirdeep2.sh so that mature_mirbase_cnidarian_T.fa is the input fasta file. Submitted batch job 292458. Failed. The report log file is telling me that I need to put none none before the -t flag. Changed that and resubmitted job. Submitted batch job 292459. Hooray appears to be running!
Sam White talked to Azenta (who did the sequencing for my project) and they recommended using Trimmomatic for trimming the small RNA reads. They also recommended tossing out read 2 and only using read 1 for analysis. I concatenate and collapse the reads anyway, so that shouldn’t matter too much. Sam will get back to me about trimming info/code from Azenta soon.
20240116
While I wait for Sam to get back to me about the trimming info, I’m going to run a mirdeep2 test on another sample (AST-2000). Unzip the files first.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
gunzip AST-2000_R1_001.fastq.*
Next, go to scripts folder and modify test_cat_collapse.sh so that the sample is AST-2000. Submitted batch job 292595. Took about 20 mins.
head sed.collapse.cat.AST-2000.fastq
>seq_1_x719979
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_2_x647300
GAGAATTCTACCACTGAACCACCAGTGC
>seq_3_x228711
GCACTGTGGTTCAGTGGTAGAATTCTC
>seq_4_x206224
GAGAATTCTACCACTGAACCACAGTGC
>seq_5_x161452
GCACTGGTGGTTCAGTGGTAGAATTCT
zgrep -c ">" sed.collapse.cat.AST-2000.fastq
10773578
Remove any sequences that are <17 nts.
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2000.fastq"
output_file="17_sed.collapse.cat.AST-2000.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2000.fastq
10773578
Looks like no sequences were removed. Now let’s attempt the mirdeep2 run with AST-2000. Map reads to genome first.
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2000.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s 20240116_reads_collapsed.fa -t 20240116_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_5771
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 34857708 5888651 28969057 16.893 83.107
seq: 34857708 5888651 28969057 16.893 83.107
Higher mapping % than AST-1065. Let’s look at the files produced.
head 20240116_reads_collapsed.fa
>seq_1_x719979
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_2_x647300
GAGAATTCTACCACTGAACCACCAGTGC
>seq_3_x228711
GCACTGTGGTTCAGTGGTAGAATTCTC
>seq_4_x206224
GAGAATTCTACCACTGAACCACAGTGC
>seq_5_x161452
GCACTGGTGGTTCAGTGGTAGAATTCT
zgrep -c ">" 20240116_reads_collapsed.fa
10773578
head 20240116_reads_collapsed_vs_genome.arf
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_27_x28319 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 20610 20639 tccgacactcagacagacatgctcctggga + mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_27_x28319 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 20240116_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
1016532
Edit the test_mirdeep2.sh script to contain info for AST-2000
#!/bin/bash -i
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/lncRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on test sample AST-2000" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2000.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/20240116_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for test sample AST-2000" $(date)
conda deactivate
Submitted batch job 292597. This took about 7 hours to run.
I then compared the AST-1065 and AST-2000 samples in R to see if there were any overlapping sequences (code here). I found 21 unique overlapping sequences between the two samples! Exciting.
20240119
Hollie recommended that I compare the 25 bp vs 30 bp to see what the outcome was (ie did one trim length yield more miRNAs than the other). First, I need to prep the AST-2000 25bp sample for mirdeep2.
Unzip the files.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar_25bp
gunzip trim.AST-2000*
Next, go to scripts folder and modify test_cat_collapse.sh so that the sample is AST-2000 from the flexbar 25 bp folder. Submitted batch job 293019. Took about 15 mins
head sed.collapse.cat.AST-2000_25bp.fastq
>seq_1_x1075819
GCACTGGTGGTTCAGTGGTAGAATT
>seq_2_x673045
GAGAATTCTACCACTGAACCACCAG
>seq_3_x339694
GCACTGTGGTTCAGTGGTAGAATTC
>seq_4_x212925
GAGAATTCTACCACTGAACCACAGT
>seq_5_x150098
AGAATTCTACCACTGAACCACCAGT
zgrep -c ">" sed.collapse.cat.AST-2000_25bp.fastq
9235930
Remove any seqs that are <17 nts.
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2000_25bp.fastq"
output_file="17_sed.collapse.cat.AST-2000_25bp.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2000_25bp.fastq
9235930
No reads removed. Now map the reads to the genome using mirdeep2
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar_25bp/17_sed.collapse.cat.AST-2000_25bp.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s 20240119_reads_collapsed.fa -t 20240119_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_51907
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 34857708 6018696 28839012 17.266 82.734
seq: 34857708 6018696 28839012 17.266 82.734
Slightly higher mapping than the 30bp AST-2000. Let’s look at the files
head 20240119_reads_collapsed.fa
>seq_1_x1075819
GCACTGGTGGTTCAGTGGTAGAATT
>seq_2_x673045
GAGAATTCTACCACTGAACCACCAG
>seq_3_x339694
GCACTGTGGTTCAGTGGTAGAATTC
>seq_4_x212925
GAGAATTCTACCACTGAACCACAGT
>seq_5_x150098
AGAATTCTACCACTGAACCACCAGT
zgrep -c ">" 20240119_reads_collapsed.fa
9235930
head 20240119_reads_collapsed_vs_genome.arf
seq_6_x145559 25 1 25 aacttttgacggtggatctcttggc chromosome_2 25 20741 20765 aacttttgacggtggatctcttggc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x145559 25 1 25 aacttttgacggtggatctcttggc chromosome_2 25 31485 31509 aacttttgacggtggatctcttggc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x145559 25 1 25 aacttttgacggtggatctcttggc chromosome_2 25 42328 42352 aacttttgacggtggatctcttggc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x145559 25 1 25 aacttttgacggtggatctcttggc chromosome_2 25 53077 53101 aacttttgacggtggatctcttggc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x42856 25 1 25 tgcgtgagccaagagatccaccgtc chromosome_2 25 42321 42345 tgcgtgagccaagagatccaccgtc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x42856 25 1 25 tgcgtgagccaagagatccaccgtc chromosome_2 25 53070 53094 tgcgtgagccaagagatccaccgtc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x42856 25 1 25 tgcgtgagccaagagatccaccgtc chromosome_2 25 31478 31502 tgcgtgagccaagagatccaccgtc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x42856 25 1 25 tgcgtgagccaagagatccaccgtc chromosome_2 25 20734 20758 tgcgtgagccaagagatccaccgtc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_27_x32954 25 1 25 tccgacactcagacagacatgctcc chromosome_2 25 42197 42221 tccgacactcagacagacatgctcc mmmmmmmmmmmmmmmmmmmmmmmmm
seq_27_x32954 25 1 25 tccgacactcagacagacatgctcc chromosome_2 25 52946 52970 tccgacactcagacagacatgctcc mmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 20240119_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
792395
Less reads collapsed vs genome for the 25 bp than the 30 bp AST-2000 sample.
Edit the test_mirdeep2.sh script to contain info for AST-2000 25bp
#!/bin/bash -i
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on test sample AST-2000 trimmed to 25bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar_25bp/17_sed.collapse.cat.AST-2000_25bp.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/20240119_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for test sample AST-2000 trimmed to 25bp" $(date)
conda deactivate
Submitted batch job 293022
20240121
Took about 16 hours to run. Now doing to download the csv and compare the AST-2000 25 vs 30 bp results.
AST-2000 25 bp
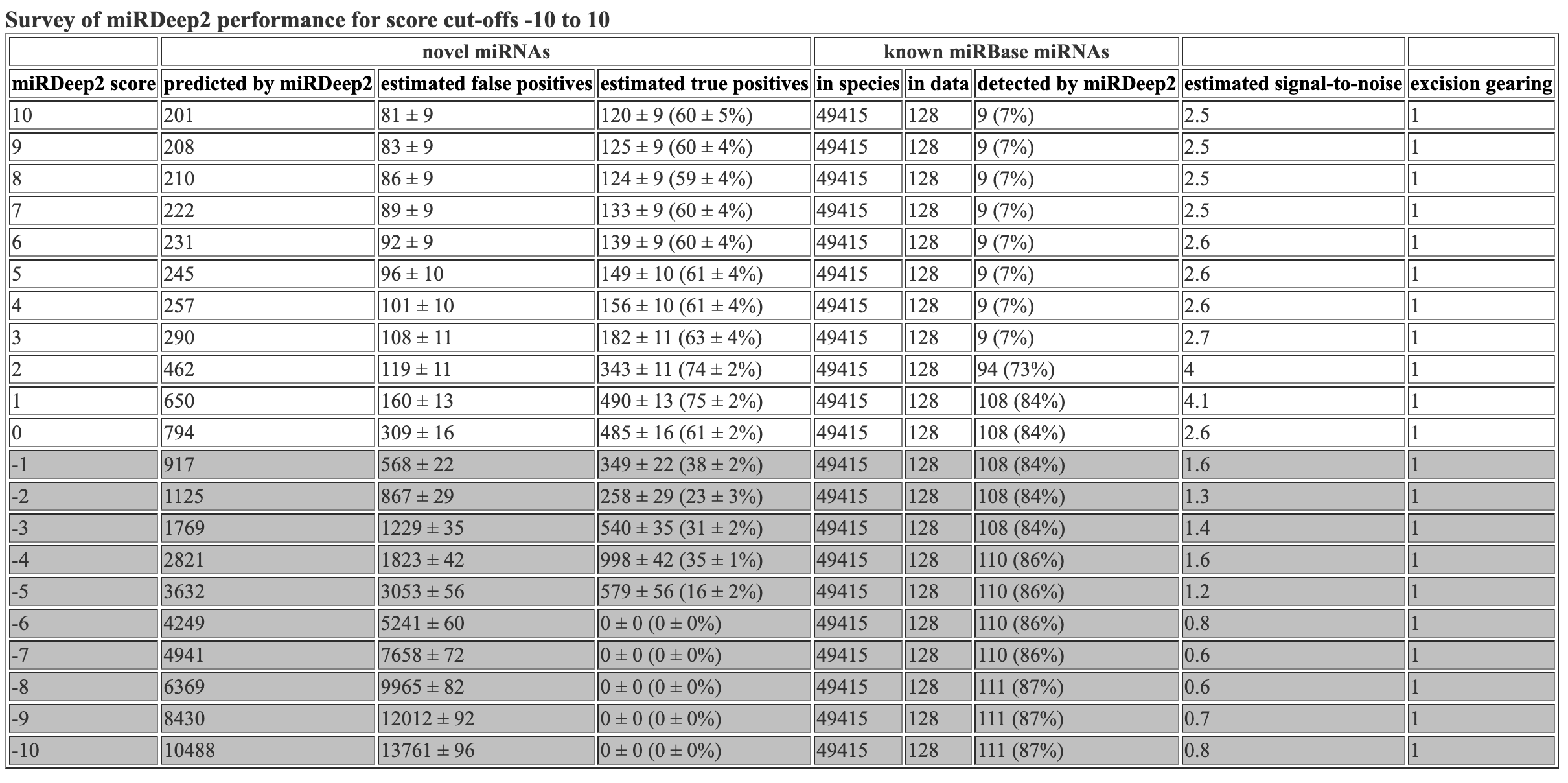
AST-2000 30 bp
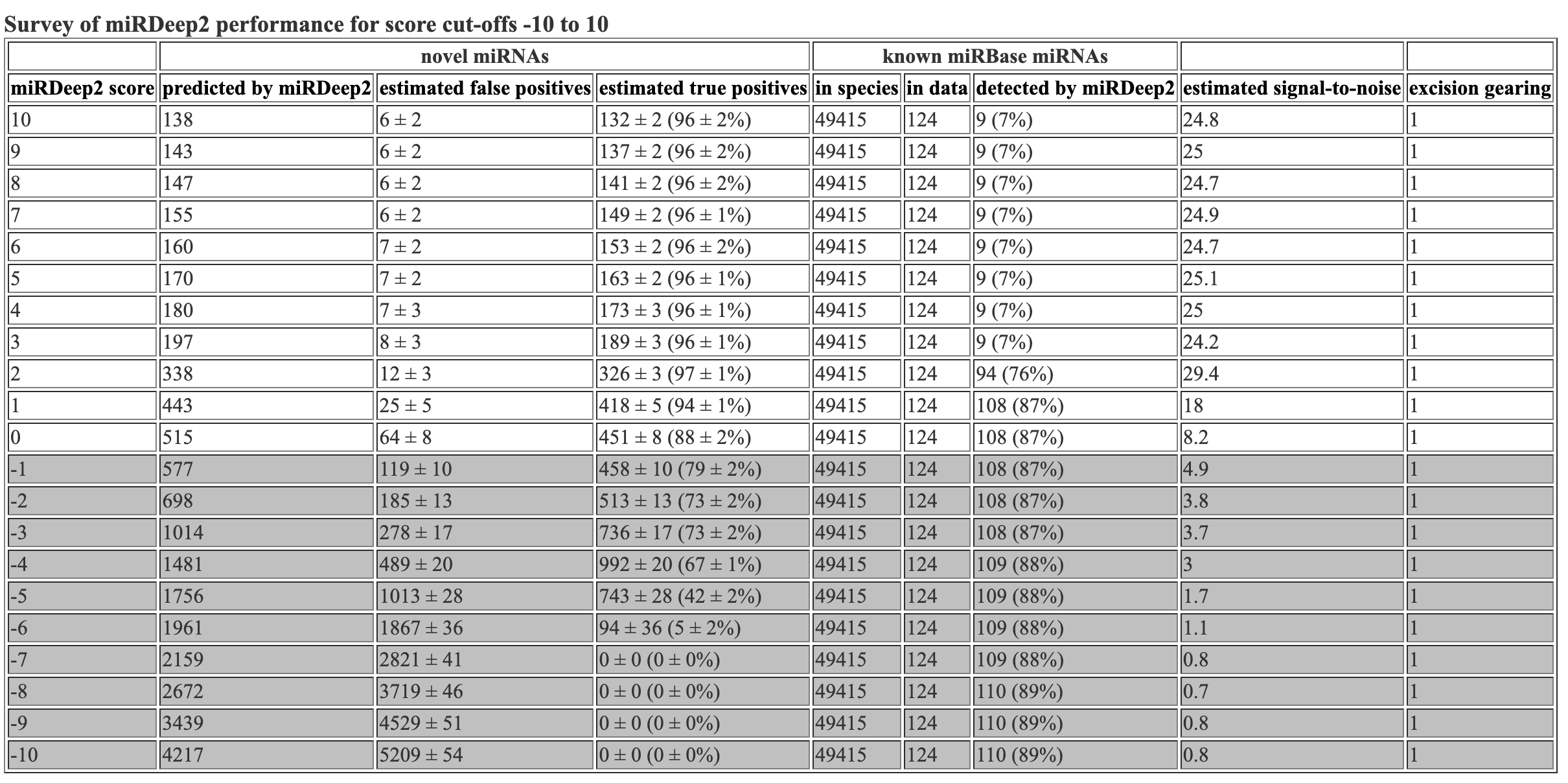
The 25bp trimming had more miRNAs identified but more false positives. There were 111 unique sews when comparing 25bp v 30bp AST-2000. I think I am going to move forward with the 30bp trimming.
Yay!!!!! Okay well now I can do the rest of the mirdeep2 runs for all the samples. Idk how I feel about putting all the samples in a loop because I don’t want them to overwrite one another. I might do a separate script for each sample…If I do that, I’ll need to make separate scripts for the mirdeep2 itself.
In /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar, gunzip all fastq files.
I can write a single script to concatenate (with cat command) and collapse reads (with fastx_collapser from the fastx toolkit. In scripts folder: nano cat_collapse.sh.
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
echo "Concatenate and collapse smRNA reads" $(date)
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
samples=$(ls *_R1_001.fastq.gz_1.fastq | sed 's/\(.*\)_R1_001.fastq.gz_1.fastq/\1/')
for sample in $samples
do
# Concatenate paired-end reads
cat "${sample}_R1_001.fastq.gz_1.fastq" "${sample}_R1_001.fastq.gz_2.fastq" > "cat.${sample}.fastq"
echo "${sample} reads are concatenated into one file per sample" $(date)
# Collapse concatenated reads
fastx_collapser -v -i "cat.${sample}.fastq" -o "collapse.cat.${sample}.fastq"
echo "${sample} reads collapsed" $(date)
done
Submitted batch job 293086
20240122
All files are now concatenated and collapsed. I need to now prep the sequence IDs for mirdeep2 with sed. I should’ve put this in the script. I’m also going to remove any reads <17nts.
AST-1065
sed '/^>/ s/-/_x/g' collapse.cat.AST-1065.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1065.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1065.fastq"
output_file="17_sed.collapse.cat.AST-1065.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1065.fastq
11979585
Not doing AST-1105 bc of poor QC and mapping
AST-1147
sed '/^>/ s/-/_x/g' collapse.cat.AST-1147.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1147.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1147.fastq"
output_file="17_sed.collapse.cat.AST-1147.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1147.fastq
18025765
AST-1412
sed '/^>/ s/-/_x/g' collapse.cat.AST-1412.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1412.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1412.fastq"
output_file="17_sed.collapse.cat.AST-1412.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1412.fastq
11251691
AST-1560
sed '/^>/ s/-/_x/g' collapse.cat.AST-1560.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1560.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1560.fastq"
output_file="17_sed.collapse.cat.AST-1560.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1560.fastq
11702120
AST-1567
sed '/^>/ s/-/_x/g' collapse.cat.AST-1567.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1567.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1567.fastq"
output_file="17_sed.collapse.cat.AST-1567.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1567.fastq
9871465
AST-1617
sed '/^>/ s/-/_x/g' collapse.cat.AST-1617.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1617.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1617.fastq"
output_file="17_sed.collapse.cat.AST-1617.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1617.fastq
8081542
AST-1722
sed '/^>/ s/-/_x/g' collapse.cat.AST-1722.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-1722.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-1722.fastq"
output_file="17_sed.collapse.cat.AST-1722.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-1722.fastq
7574236
AST-2000
sed '/^>/ s/-/_x/g' collapse.cat.AST-2000.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2000.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2000.fastq"
output_file="17_sed.collapse.cat.AST-2000.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2000.fastq
10773578
AST-2007
sed '/^>/ s/-/_x/g' collapse.cat.AST-2007.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2007.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2007.fastq"
output_file="17_sed.collapse.cat.AST-2007.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2007.fastq
8653745
AST-2302
sed '/^>/ s/-/_x/g' collapse.cat.AST-2302.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2302.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2302.fastq"
output_file="17_sed.collapse.cat.AST-2302.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2302.fastq
11391780
AST-2360
sed '/^>/ s/-/_x/g' collapse.cat.AST-2360.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2360.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2360.fastq"
output_file="17_sed.collapse.cat.AST-2360.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2360.fastq
9737775
AST-2398
sed '/^>/ s/-/_x/g' collapse.cat.AST-2398.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2398.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2398.fastq"
output_file="17_sed.collapse.cat.AST-2398.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2398.fastq
9507430
AST-2404
sed '/^>/ s/-/_x/g' collapse.cat.AST-2404.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2404.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2404.fastq"
output_file="17_sed.collapse.cat.AST-2404.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2404.fastq
11599063
AST-2412
sed '/^>/ s/-/_x/g' collapse.cat.AST-2412.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2412.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2412.fastq"
output_file="17_sed.collapse.cat.AST-2412.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2412.fastq
10485205
AST-2512
sed '/^>/ s/-/_x/g' collapse.cat.AST-2512.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2512.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2512.fastq"
output_file="17_sed.collapse.cat.AST-2512.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2512.fastq
9710109
AST-2523
sed '/^>/ s/-/_x/g' collapse.cat.AST-2523.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2523.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2523.fastq"
output_file="17_sed.collapse.cat.AST-2523.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2523.fastq
11748402
AST-2563
sed '/^>/ s/-/_x/g' collapse.cat.AST-2563.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2563.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2563.fastq"
output_file="17_sed.collapse.cat.AST-2563.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2563.fastq
11689234
AST-2729
sed '/^>/ s/-/_x/g' collapse.cat.AST-2729.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2729.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2729.fastq"
output_file="17_sed.collapse.cat.AST-2729.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2729.fastq
10703305
AST-2755
sed '/^>/ s/-/_x/g' collapse.cat.AST-2755.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.AST-2755.fastq
#!/bin/bash
# Define the input and output files
input_file="sed.collapse.cat.AST-2755.fastq"
output_file="17_sed.collapse.cat.AST-2755.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.AST-2755.fastq
10834414
Okay everything is prepped! I’m going to remove the extra files made (ie cat, collapse, sed iterations of the file). Going back to the main folder, I’m going to make a mirdeep2 folder that I will run all of my mirdeep2 code in so that the output is all in one place.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA
mkdir mirdeep2
cd mirdeep2
mkdir AST-1065 AST-1147 AST-1412 AST-1560 AST-1567 AST-1617 AST-1722 AST-2000 AST-2007 AST-2302 AST-2360 AST-2398 AST-2404 AST-2412 AST-2512 AST-2523 AST-2563 AST-2729 AST-2755
Now let’s run mirdeep2 on all of the samples! I am paranoid that if I run the script in a loop for all the samples, something is going to overwrite. Therefore, I am going to run each sample individually.
AST-1065
cd AST-1065
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1065_reads_collapsed.fa -t AST-1065_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_51087
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 35658222 3275049 32383173 9.185 90.815
seq: 35658222 3275049 32383173 9.185 90.815
Look at the mapping results
head AST-1065_reads_collapsed.fa
>seq_1_x357414
TGGTCTATGGTGTAACTGGCAACACGTCTG
>seq_2_x138955
ACAGACGTGTTGCCAGTTACACCATAGACC
>seq_3_x125294
AACAGACGTGTTGCCAGTTACACCATAGAC
>seq_4_x98253
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_5_x87633
TTCCACTTCAGAGAAAAGATTTTCA
zgrep -c ">" AST-1065_reads_collapsed.fa
11979585
head AST-1065_reads_collapsed_vs_genome.arf
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x81424 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 27510 27539 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 49105 49134 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 38360 38389 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x38316 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 879106 879135 acaaatcttagaacaaaggcttaatctcag - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_25_x32759 30 1 30 ttgctacgatcttctgagattaagcctttg chromosome_2 30 879093 879122 ttgctacgatcttctgagattaagcctttg + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_25_x32759 30 1 30 ttgctacgatcttctgagattaagcctttg chromosome_2 30 38373 38402 ttgctacgatcttctgagattaagcctttg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-1065_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
523747
conda deactivate
Run mirdeep2. nano AST-1065_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1065 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065/AST-1065_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1065 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293148
AST-1147
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1147
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1147.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1147_reads_collapsed.fa -t AST-1147_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_53573
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 80830448 23638038 57192410 29.244 70.756
seq: 80830448 23638038 57192410 29.244 70.756
Look at the mapping results
head AST-1147_reads_collapsed.fa
>seq_1_x3434945
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x1817679
GGCGAGAATTCTACCACTGAACCACCAGTG
>seq_3_x926642
AGCGAGAATTCTACCACTGAACCACCAGTG
>seq_4_x602263
GCACTGTGGTTCAGTGGTAGAATTCTCGCC
>seq_5_x475440
GGCGAGAATTCTACCACTGAACCACAGTGC
zgrep -c ">" AST-1147_reads_collapsed.fa
18025765
head AST-1147_reads_collapsed_vs_genome.arf
seq_6_x427578 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x427578 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x427578 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x427578 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_14_x202590 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_14_x202590 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_14_x202590 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_14_x202590 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x127492 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 879106 879135 acaaatcttagaacaaaggcttaatctcag - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x127492 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 38360 38389 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-1147_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
3227088
conda deactivate
Run mirdeep2. nano AST-1147_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1147
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1147 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1147.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1147/AST-1147_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1147 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293149
AST-1412
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1412
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1412.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1412_reads_collapsed.fa -t AST-1412_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_56487
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 32559110 3100518 29458592 9.523 90.477
seq: 32559110 3100518 29458592 9.523 90.477
Look at the mapping results
head AST-1412_reads_collapsed.fa
>seq_1_x137015
GGAAGAGCACACGTCTGAACTCCAGTCACT
>seq_2_x100202
AACTTTTGACGGTGGATCTCTTGGCTCACG
>seq_3_x94707
TCGGACTGTAGAACTCTGAACGTGTAGATC
>seq_4_x78147
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_5_x73890
TACTGGATAACTAAGGGAAAGTTTGGCTAA
zgrep -c ">" AST-1412_reads_collapsed.fa
11251691
head AST-1412_reads_collapsed_vs_genome.arf
seq_2_x100202 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_2_x100202 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_2_x100202 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_2_x100202 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x39468 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x39468 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x39468 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_19_x39468 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x31213 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x31213 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-1412_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
599077
conda deactivate
Run mirdeep2. nano AST-1412_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1412
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1412 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1412.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1412/AST-1412_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1412 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293150
AST-1560
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1560
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1560.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1560_reads_collapsed.fa -t AST-1560_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_58565
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 35654048 767007 34887041 2.151 97.849
seq: 35654048 767007 34887041 2.151 97.849
Look at the mapping results
head AST-1560_reads_collapsed.fa
>seq_1_x404539
GACTTTGTAGCATAGGTAAGGTTAGTGCAT
>seq_2_x272137
TCAGATGCACTAACCTTACCTATGCTACAA
>seq_3_x78722
GGAAGAGCACACGTCTGAACTCCAGTCACG
>seq_4_x58838
TCGGACTGTAGAACTCTGAACGTGTAGATC
>seq_5_x46651
GCACTGATGGTTCAGTGGTAGAATTCTCGC
zgrep -c ">" AST-1560_reads_collapsed.fa
11702120
head AST-1560_reads_collapsed_vs_genome.arf
seq_22_x19193 30 1 30 caggtctgtgatgcccttagatgtccgggg chromosome_2 30 32082 32111 caggtctgtgatgcccttagatgtacaggg - 2 mmmmmmmmmmmmmmmmmmmmmmmmMmMmmm
seq_22_x19193 30 1 30 caggtctgtgatgcccttagatgtccgggg chromosome_2 30 42925 42954 caggtctgtgatgcccttagatgtacaggg - 2 mmmmmmmmmmmmmmmmmmmmmmmmMmMmmm
seq_22_x19193 30 1 30 caggtctgtgatgcccttagatgtccgggg chromosome_2 30 21338 21367 caggtctgtgatgcccttagatgtacaggg - 2 mmmmmmmmmmmmmmmmmmmmmmmmMmMmmm
seq_43_x14818 30 1 30 acaggtctgtgatgcccttagatgtccggg chromosome_2 30 32083 32112 acaggtctgtgatgcccttagatgtacagg - 2 mmmmmmmmmmmmmmmmmmmmmmmmmMmMmm
seq_43_x14818 30 1 30 acaggtctgtgatgcccttagatgtccggg chromosome_2 30 42926 42955 acaggtctgtgatgcccttagatgtacagg - 2 mmmmmmmmmmmmmmmmmmmmmmmmmMmMmm
seq_43_x14818 30 1 30 acaggtctgtgatgcccttagatgtccggg chromosome_2 30 21339 21368 acaggtctgtgatgcccttagatgtacagg - 2 mmmmmmmmmmmmmmmmmmmmmmmmmMmMmm
seq_72_x11510 30 1 30 aggtctgtgatgcccttagatgtccggggc chromosome_2 30 32081 32110 aggtctgtgatgcccttagatgtacagggc - 2 mmmmmmmmmmmmmmmmmmmmmmmMmMmmmm
seq_72_x11510 30 1 30 aggtctgtgatgcccttagatgtccggggc chromosome_2 30 42924 42953 aggtctgtgatgcccttagatgtacagggc - 2 mmmmmmmmmmmmmmmmmmmmmmmMmMmmmm
seq_72_x11510 30 1 30 aggtctgtgatgcccttagatgtccggggc chromosome_2 30 21337 21366 aggtctgtgatgcccttagatgtacagggc - 2 mmmmmmmmmmmmmmmmmmmmmmmMmMmmmm
seq_87_x10194 30 1 30 agccaggaatcctaaccgctagaccatctg chromosome_12 30 41900395 41900424 agccaggaatcctaaccgctagaccatttg - 1 mmmmmmmmmmmmmmmmmmmmmmmmmmmMmm
cut -f1 AST-1560_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
123911
conda deactivate
Run mirdeep2. nano AST-1560_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1560
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1560 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1560.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1560/AST-1560_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1560 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293152
AST-1567
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1567
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1567.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1567_reads_collapsed.fa -t AST-1567_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_12237
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33222794 3149689 30073105 9.481 90.519
seq: 33222794 3149689 30073105 9.481 90.519
Look at the mapping results
head AST-1567_reads_collapsed.fa
>seq_1_x330204
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x226911
GCACTGATGGTTCAGTGGTAGAATTCTCGC
>seq_3_x157338
GGCGAGAATTCTACCACTGAACCACCAGTG
>seq_4_x98181
AGCGAGAATTCTACCACTGAACCACCAGTG
>seq_5_x77909
GGAAGAGCACACGTCTGAACTCCAGTCACA
zgrep -c ">" AST-1567_reads_collapsed.fa
9871465
head AST-1567_reads_collapsed_vs_genome.arf
seq_9_x59542 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x59542 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x59542 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x59542 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_24_x25786 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_24_x25786 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_24_x25786 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_24_x25786 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_37_x16700 29 1 29 aatggataaccctcaaccgtccggacctc chromosome_14 29 6295369 6295397 aatggataaccctcaaccgtccggacctc - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_54_x12863 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-1567_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
569922
conda deactivate
Run mirdeep2. nano AST-1567_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1567
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1567 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1567.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1567/AST-1567_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1567 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293153
I’m going to call it quits for today. 3 samples are running through mirdeep2 now, and 2 samples are pending on the server.
20240123
Last night/early this morning, AST-1054, AST-1412, AST-1560, and AST-1567 all finished running mirdeep2. I downloaded the html and csv ouput files to my computer. I’m going to continue
AST-1617
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1617
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1617.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1617_reads_collapsed.fa -t AST-1617_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_11212
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 32155434 7494814 24660620 23.308 76.692
seq: 32155434 7494814 24660620 23.308 76.692
Look at the mapping results
head AST-1617_reads_collapsed.fa
>seq_1_x1583958
TAAGACTATGATTATATGCAGCTTCTTGCA
>seq_2_x1378737
ATTGGTTTCGAGATGCAAGAAGCTGCATAT
>seq_3_x447663
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_4_x368633
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_5_x329309
GAGAATTCTACCACTGAACCACCAGTGC
zgrep -c ">" AST-1617_reads_collapsed.fa
8081542
head AST-1617_reads_collapsed_vs_genome.arf
seq_7_x232409 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x232409 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x232409 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x232409 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x108899 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x108899 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x108899 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x108899 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_30_x33370 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_30_x33370 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 20610 20639 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-1617_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
1368740
conda deactivate
Run mirdeep2. nano AST-1617_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1617
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1617 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1617.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1617/AST-1617_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1617 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293162
AST-1722
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1722
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1722.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-1722_reads_collapsed.fa -t AST-1722_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_13688
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 32860442 11326758 21533684 34.469 65.531
seq: 32860442 11326758 21533684 34.469 65.531
Look at the mapping results
head AST-1722_reads_collapsed.fa
>seq_1_x1663348
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x942322
GGCGAGAATTCTACCACTGAACCACCAGTG
>seq_3_x422823
AGCGAGAATTCTACCACTGAACCACCAGTG
>seq_4_x345835
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_5_x329076
GCACTGTGGTTCAGTGGTAGAATTCTCGCC
zgrep -c ">" AST-1722_reads_collapsed.fa
7574236
head AST-1722_reads_collapsed_vs_genome.arf
seq_6_x311272 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x311272 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x311272 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_6_x311272 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x140612 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x140612 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x140612 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x140612 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x123585 30 1 30 cgagctgttcttcctcgcaaagactgtgtg chromosome_2 30 32824 32853 cgagctgttcttcctcgcaaagactgtgtg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x123585 30 1 30 cgagctgttcttcctcgcaaagactgtgtg chromosome_2 30 43667 43696 cgagctgttcttcctcgcaaagactgtgtg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-1722_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
1688293
conda deactivate
Run mirdeep2. nano AST-1722_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1722
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-1722 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1722.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1722/AST-1722_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-1722 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293188
AST-2000
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2000
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2000.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2000_reads_collapsed.fa -t AST-2000_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_15165
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 34857708 5888651 28969057 16.893 83.107
seq: 34857708 5888651 28969057 16.893 83.107
Look at the mapping results
head AST-2000_reads_collapsed.fa
>seq_1_x719979
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_2_x647300
GAGAATTCTACCACTGAACCACCAGTGC
>seq_3_x228711
GCACTGTGGTTCAGTGGTAGAATTCTC
>seq_4_x206224
GAGAATTCTACCACTGAACCACAGTGC
>seq_5_x161452
GCACTGGTGGTTCAGTGGTAGAATTCT
zgrep -c ">" AST-2000_reads_collapsed.fa
10773578
head AST-2000_reads_collapsed_vs_genome.arf
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_7_x119692 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_15_x41699 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_27_x28319 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 20610 20639 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_27_x28319 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2000_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
1016532
conda deactivate
Run mirdeep2. nano AST-2000_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2000
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2000 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2000.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2000/AST-2000_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2000 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293203
AST-2007
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2007
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2007.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2007_reads_collapsed.fa -t AST-2007_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_18266
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33119102 7261428 25857674 21.925 78.075
seq: 33119102 7261428 25857674 21.925 78.075
Look at the mapping results
head AST-2007_reads_collapsed.fa
>seq_1_x745390
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_2_x666548
TTCCACTTCAGAGAAAAGATTTTCA
>seq_3_x176638
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_4_x162570
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_5_x157967
GAGAATTCTACCACTGAACCACCAGTGC
zgrep -c ">" AST-2007_reads_collapsed.fa
8653745
head AST-2007_reads_collapsed_vs_genome.arf
seq_9_x90166 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x90166 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x90166 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x90166 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_11_x64281 30 1 30 agcacacacagtctttgcgaggaagaacag chromosome_2 30 22076 22105 agcacacacagtctttgcgaggaagaacag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_11_x64281 30 1 30 agcacacacagtctttgcgaggaagaacag chromosome_2 30 43663 43692 agcacacacagtctttgcgaggaagaacag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_11_x64281 30 1 30 agcacacacagtctttgcgaggaagaacag chromosome_2 30 32820 32849 agcacacacagtctttgcgaggaagaacag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x52870 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 879106 879135 acaaatcttagaacaaaggcttaatctcag - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x52870 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 38360 38389 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x52870 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 27510 27539 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2007_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
949862
conda deactivate
Run mirdeep2. nano AST-2007_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2007
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2007 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2007.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2007/AST-2007_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2007 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293252
AST-2302
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2302
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2302.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2302_reads_collapsed.fa -t AST-2302_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_35942
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33330740 5544909 27785831 16.636 83.364
seq: 33330740 5544909 27785831 16.636 83.364
Look at the mapping results
head AST-2302_reads_collapsed.fa
>seq_1_x184430
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x151792
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_3_x131568
TTCCACTTCAGAGAAAAGATTTTCA
>seq_4_x102320
GGCGAGAATTCTACCACTGAACCACCAGTG
>seq_5_x88502
GCACTGGTGGTTCAGTGGTAGAATTCTC
zgrep -c ">" AST-2302_reads_collapsed.fa
11391780
head AST-2302_reads_collapsed_vs_genome.arf
seq_8_x67603 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_8_x67603 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_8_x67603 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_8_x67603 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_21_x35069 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_21_x35069 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_21_x35069 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 20610 20639 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_21_x35069 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_30_x23771 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 38360 38389 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_30_x23771 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 27510 27539 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2302_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
1149823
conda deactivate
Run mirdeep2. nano AST-2302_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2302
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2302 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2302.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2302/AST-2302_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2302 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293254
AST-2360
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2360
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2360.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2360_reads_collapsed.fa -t AST-2360_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_39928
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33296712 4751266 28545446 14.269 85.731
seq: 33296712 4751266 28545446 14.269 85.731
Look at the mapping results
head AST-2360_reads_collapsed.fa
>seq_1_x265109
TAAGACTATGATTATATGCAGCTTCTTGCA
>seq_2_x223703
ATTGGTTTCGAGATGCAAGAAGCTGCATAT
>seq_3_x134102
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_4_x86252
AACTTTTGACGGTGGATCTCTTGGCTCACG
>seq_5_x66786
GGCGAGAATTCTACCACTGAACCACCAGTG
zgrep -c ">" AST-2360_reads_collapsed.fa
9737775
head AST-2360_reads_collapsed_vs_genome.arf
seq_4_x86252 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_4_x86252 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_4_x86252 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_4_x86252 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_22_x22715 30 1 30 aagagcgccatttgcgttcaaagattcgat chromosome_2 30 52982 53011 aagagcgccatttgcgttcaaagattcgat + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_22_x22715 30 1 30 aagagcgccatttgcgttcaaagattcgat chromosome_2 30 31390 31419 aagagcgccatttgcgttcaaagattcgat + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_22_x22715 30 1 30 aagagcgccatttgcgttcaaagattcgat chromosome_2 30 20646 20675 aagagcgccatttgcgttcaaagattcgat + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_22_x22715 30 1 30 aagagcgccatttgcgttcaaagattcgat chromosome_2 30 42233 42262 aagagcgccatttgcgttcaaagattcgat + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_24_x22365 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_24_x22365 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2360_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
464780
conda deactivate
Run mirdeep2. nano AST-2360_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2360
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2360 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2360.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2360/AST-2360_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2360 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293255
AST-2398
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2398
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2398.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2398_reads_collapsed.fa -t AST-2398_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_48437
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33576416 7946641 25629775 23.667 76.333
seq: 33576416 7946641 25629775 23.667 76.333
Look at the mapping results
head AST-2398_reads_collapsed.fa
>seq_1_x881553
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x433411
GGCGAGAATTCTACCACTGAACCACCAGTG
>seq_3_x266696
AGCGAGAATTCTACCACTGAACCACCAGTG
>seq_4_x218909
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_5_x195072
GAGAATTCTACCACTGAACCACCAGTGC
zgrep -c ">" AST-2398_reads_collapsed.fa
9507430
head AST-2398_reads_collapsed_vs_genome.arf
seq_8_x145611 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_8_x145611 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_8_x145611 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_8_x145611 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_17_x64706 29 1 29 aatggataaccctcaaccgtccggacctc chromosome_14 29 6295369 6295397 aatggataaccctcaaccgtccggacctc - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x61937 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x61937 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x61937 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_18_x61937 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_21_x38252 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 27510 27539 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2398_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
conda deactivate
Run mirdeep2. nano AST-2398_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2398
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2398 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2398.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2398/AST-2398_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2398 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293257
need to run quantifier module for samples: https://github.com/rajewsky-lab/mirdeep2/tree/master
How do I find the MFE? Is it calculated by mirdeep2 or by the quantifier module? I think it is linked to the randfold step. Need to look into this.
I looked at Gajigan & Conaco 2017 mirdeep2 pdf outputs from their supplementary materials and they got similar MFE values in their pdfs. However, in Table S5, they have MFE info that is <-25 kcal/mol. How did they calculate the MFE that mirdeep2 gave them to the MFE that was displayed in their table??
20240124
All of the mirdeep2 jobs finished last night/early this morning. Now time to start some more.
AST-2404
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2404
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2404.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2404_reads_collapsed.fa -t AST-2404_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_58495
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33425806 5062194 28363612 15.145 84.855
seq: 33425806 5062194 28363612 15.145 84.855
Look at the mapping results
head AST-2404_reads_collapsed.fa
>seq_1_x144129
GGAAGAGCACACGTCTGAACTCCAGTCACC
>seq_2_x132047
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_3_x124405
AACTTTTGACGGTGGATCTCTTGGCTCACG
>seq_4_x97061
TCGGACTGTAGAACTCTGAACGTGTAGATC
>seq_5_x94760
GCACTGATGGTTCAGTGGTAGAATTCTCGC
zgrep -c ">" AST-2404_reads_collapsed.fa
11599063
head AST-2404_reads_collapsed_vs_genome.arf
seq_3_x124405 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_3_x124405 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_3_x124405 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_3_x124405 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x51804 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x51804 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x51804 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_9_x51804 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_11_x39689 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_11_x39689 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2404_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
881575
conda deactivate
Run mirdeep2. nano AST-2404_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2404
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2404 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2404.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2404/AST-2404_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2404 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293308
AST-2412
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2412
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2412.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2412_reads_collapsed.fa -t AST-2412_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_59645
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 34977016 2244564 32732452 6.417 93.583
seq: 34977016 2244564 32732452 6.417 93.583
Look at the mapping results
head AST-2412_reads_collapsed.fa
>seq_1_x1651314
GACTTTGTAGCATAGGTAAGGTTAGTGCAT
>seq_2_x1124747
TCAGATGCACTAACCTTACCTATGCTACAA
>seq_3_x189997
CAGATGCACTAACCTTACCTATGCTACAAA
>seq_4_x98647
AACTTTTGACGGTGGATCTCTTGGCTCACG
>seq_5_x84357
ATCAGATGCACTAACCTTACCTATGCTACA
zgrep -c ">" AST-2412_reads_collapsed.fa
10485205
head AST-2412_reads_collapsed_vs_genome.arf
seq_4_x98647 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_4_x98647 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_4_x98647 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_4_x98647 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x44893 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x44893 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x44893 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x44893 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_33_x19915 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_33_x19915 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2412_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
415100
conda deactivate
Run mirdeep2. nano AST-2412_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2412
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2412 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2412.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2412/AST-2412_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2412 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293309
AST-2512
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2512
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2512.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2512_reads_collapsed.fa -t AST-2512_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_60774
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 32531432 1232618 31298814 3.789 96.211
seq: 32531432 1232618 31298814 3.789 96.211
Look at the mapping results
head AST-2512_reads_collapsed.fa
>seq_1_x1229410
AAATACAAATCGTTCAGGTATTAGGAGTGA
>seq_2_x900725
AGCTCACTCCTAATACCTGAACGATTTGTA
>seq_3_x539793
AGATGGAATTGTAGCATG
>seq_4_x488884
CATGCTACAATTCCATCT
>seq_5_x298053
ACTGGATAACTAAGGGAAAGTTTGGCTAAT
zgrep -c ">" AST-2512_reads_collapsed.fa
9710109
head AST-2512_reads_collapsed_vs_genome.arf
seq_20_x37501 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x37501 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x37501 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_20_x37501 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_51_x16071 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 42321 42350 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_51_x16071 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 53070 53099 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_51_x16071 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_51_x16071 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_76_x10820 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_76_x10820 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2512_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
237458
conda deactivate
Run mirdeep2. nano AST-2512_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2512
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2512 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2512.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2512/AST-2512_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2512 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293310
AST-2523
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2523
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2523.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2523_reads_collapsed.fa -t AST-2523_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_61964
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 33990530 4117469 29873061 12.114 87.886
seq: 33990530 4117469 29873061 12.114 87.886
Look at the mapping results
head AST-2523_reads_collapsed.fa
zgrep -c ">" AST-2523_reads_collapsed.fa
head AST-2523_reads_collapsed_vs_genome.arf
cut -f1 AST-2523_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
conda deactivate
Run mirdeep2. nano AST-2523_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2523
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2523 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2523.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2523/AST-2523_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2523 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293319
I want to try running the quantifier module. on the mirdeep2 github, it says that the input should be:
- A FASTA file with precursor sequences,
- A FASTA file with mature miRNA sequences,
- A FASTA file with deep sequencing reads, and
- Optionally a FASTA file with star sequences and the 3 letter code of the species of interest.
I need to create fasta files with the known and novel mature and precursor sequences. Going to test this on AST-1065 first.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065/mirna_results_22_01_2024_t_17_11_47
cat known_mature_22_01_2024_t_17_11_47_score-50_to_na.fa novel_mature_22_01_2024_t_17_11_47_score-50_to_na.fa > AST-1065-known_novel_mature.fa
cat known_pres_22_01_2024_t_17_11_47_score-50_to_na.fa novel_pres_22_01_2024_t_17_11_47_score-50_to_na.fa > AST-1065-known_novel_pres.fa
Now I have files with the known and novel mature and precursor sequences. I hope I am correct in my interpretation of the mature and precursors seqs coming from the Time to run quantifier!
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065
conda activate /data/putnamlab/mirdeep2
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065/mirna_results_22_01_2024_t_17_11_47/AST-1065-known_novel_pres.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065/mirna_results_22_01_2024_t_17_11_47/AST-1065-known_novel_mature.fa -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq
getting samples and corresponding read numbers
Converting input files
building bowtie index
mapping mature sequences against index
mapping read sequences against index
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 35658222 69287 35588935 0.194 99.806
seq: 35658222 69287 35588935 0.194 99.806
analyzing data
326 mature mappings to precursors
Expressed miRNAs are written to expression_analyses/expression_analyses_1706128068/miRNA_expressed.csv
not expressed miRNAs are written to expression_analyses/expression_analyses_1706128068/miRNA_not_expressed.csv
Creating miRBase.mrd file
Mapped READS readin - DONE
Very low mapping…but 326 mature mapping to precursors is good because I had 326 mature miRNA seqs to begin with. Looking at the output, there are different read counts produced from the mirdeep2.pl module and the quantifier.pl module. I’m still not sure if I’m supposed to use the mirbase mature and precursor seqs or the ones produced from the mirdeep output…Let’s try running the quantifier module again using the mirbase info instead. I’m using the cnidarian mature miRNA fasta that I created a while ago. I downloaded the hairpins.fa file from mirbase to use as the fasta for the precursor sequences.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-1065
conda activate /data/putnamlab/mirdeep2
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/hairpin.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-1065.fastq
getting samples and corresponding read numbers
Converting input files
building bowtie index
mapping mature sequences against index
mapping read sequences against index
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 35658222 168104 35490118 0.471 99.529
seq: 35658222 168104 35490118 0.471 99.529
analyzing data
53298 mature mappings to precursors
Expressed miRNAs are written to expression_analyses/expression_analyses_1706129674/miRNA_expressed.csv
not expressed miRNAs are written to expression_analyses/expression_analyses_1706129674/miRNA_not_expressed.csv
Creating miRBase.mrd file
Mapped READS readin - DONE
Instead of creating pdfs, I am getting this: Negative repeat count does nothing at /data/putnamlab/mirdeep2/bin/quantifier.pl line 1312, <IN> line 122912. Not sure what it means but it makes me think that I should use the former script for quantifying. Okay yes I should use the other script. The expressed miRNA counts was just the IDs from mirbase, so that doesn’t help me very much
Also tried to figure out how to calculate MFE based on the supplemental files from Gajigan & Conaco 2017 paper. In the supplement, they inlcuded their PDF output with the MFE scores (which are similar to mine) and their supplemental table with MFE scores in kcal/mol-1, which were all -18 or lower. I plotted them against one another (x axis was MFE in kcal/mol-1, y axis was MFE from pdf) and got the slope of that line (y = -0.0437*x + 0.73, R-squared = 0.604). Then I rearranged the equation to solve for y (x = (0.73-y)/0.0437). I used the MFE pdf value as the y and attempted to re-calculate the MFE in kcal/mol. The equation did not exactly capture MFE, as the output was not the same as the MFE values reported. Here is the google sheet where I did those calculations. HOW TO CALCULATE MFE??????? This paper and this paper may help. Will look into it tomorrow.
20240129
RE my notes above, I did not look at them but it is still on my list of things to do. First, I’m going to prioritize finishing running mirdeep2 for all samples.
AST-2563
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2563
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2563.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2563_reads_collapsed.fa -t AST-2563_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_53481
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 34046004 3503553 30542451 10.291 89.709
seq: 34046004 3503553 30542451 10.291 89.709
Look at the mapping results
head AST-2563_reads_collapsed.fa
>seq_1_x117943
ATGCGTAGTGGAATACTCTGGAAAGTGT
>seq_2_x105383
ACACTTTCCAGAGTATTCCACTACGCAT
>seq_3_x69250
AACTTTTGACGGTGGATCTCTTGGCTCACG
>seq_4_x59605
GGAAGAGCACACGTCTGAACTCCAGTCACA
>seq_5_x56451
AGAAATGTGTGTAGCTGAGCAGTACTAATT
zgrep -c ">" AST-2563_reads_collapsed.fa
11689234
head AST-2563_reads_collapsed_vs_genome.arf
seq_3_x69250 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_3_x69250 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_3_x69250 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_3_x69250 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x27176 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x27176 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x27176 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_16_x27176 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 20610 20639 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_26_x22828 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_26_x22828 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2563_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
671528
conda deactivate
Run mirdeep2. nano AST-2563_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2563
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2563 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2563.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2563/AST-2563_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2563 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293787
AST-2729
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2729
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2729.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2729_reads_collapsed.fa -t AST-2729_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_54539
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 34243738 1423780 32819958 4.158 95.842
seq: 34243738 1423780 32819958 4.158 95.842
Look at the mapping results
head AST-2729_reads_collapsed.fa
>seq_1_x228146
GCACTGATGGTTCAGTGGTAGAATTCTCGC
>seq_2_x188816
GGAAGAGCACACGTCTGAACTCCAGTCACC
>seq_3_x137884
TCGGACTGTAGAACTCTGAACGTGTAGATC
>seq_4_x104317
AACTCTAAGCGGTGGATCACTCGGCTCGTG
>seq_5_x86774
ACACACGAGCCGAGTGATCCACCGCTTAGA
zgrep -c ">" AST-2729_reads_collapsed.fa
10703305
head AST-2729_reads_collapsed_vs_genome.arf
seq_36_x27457 30 1 30 tggctccccggcggggaatcgaaccccggt chromosome_14 30 42692589 42692618 tggctccccggcggggaatcgaaccccggt - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_59_x18783 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_59_x18783 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_59_x18783 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_59_x18783 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_94_x13183 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 52946 52975 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_94_x13183 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 31354 31383 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_94_x13183 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 20610 20639 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_94_x13183 30 1 30 tccgacactcagacagacatgctcctggga chromosome_2 30 42197 42226 tccgacactcagacagacatgctcctggga + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_201_x7329 30 1 30 tggctccccggcggggaaatgaaccccggt chromosome_14 30 42692589 42692618 tggctccccggcggggaaaggaaccccggt - 2 mmmmmmmmmmmmmmmmmmMMmmmmmmmmmm
cut -f1 AST-2729_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
299482
conda deactivate
Run mirdeep2. nano AST-2729_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2729
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2729 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2729.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2729/AST-2729_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2729 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293788
AST-2755
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2755
Run mapping step
conda activate /data/putnamlab/mirdeep2
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2755.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s AST-2755_reads_collapsed.fa -t AST-2755_reads_collapsed_vs_genome.arf -v
discarding short reads
mapping reads to genome index
trimming unmapped nts in the 3' ends
Log file for this run is in mapper_logs and called mapper.log_55943
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 32539646 3255098 29284548 10.003 89.997
seq: 32539646 3255098 29284548 10.003 89.997
Look at the mapping results
head AST-2755_reads_collapsed.fa
>seq_1_x282696
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x165033
GGCGAGAATTCTACCACTGAACCACCAGTG
>seq_3_x76982
AGCGAGAATTCTACCACTGAACCACCAGTG
>seq_4_x73794
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_5_x66624
TTCCACTTCAGAGAAAAGATTTTCA
zgrep -c ">" AST-2755_reads_collapsed.fa
10834414
head AST-2755_reads_collapsed_vs_genome.arf
seq_12_x49088 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 42323 42352 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x49088 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 53072 53101 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x49088 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 20736 20765 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_12_x49088 30 1 30 aacttttgacggtggatctcttggctcacg chromosome_2 30 31480 31509 aacttttgacggtggatctcttggctcacg - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_35_x24220 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 879106 879135 acaaatcttagaacaaaggcttaatctcag - 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_35_x24220 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 38360 38389 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_35_x24220 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 27510 27539 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_35_x24220 30 1 30 acaaatcttagaacaaaggcttaatctcag chromosome_2 30 49105 49134 acaaatcttagaacaaaggcttaatctcag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_40_x21090 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 31478 31507 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
seq_40_x21090 30 1 30 tgcgtgagccaagagatccaccgtcaaaag chromosome_2 30 20734 20763 tgcgtgagccaagagatccaccgtcaaaag + 0 mmmmmmmmmmmmmmmmmmmmmmmmmmmmmm
cut -f1 AST-2755_reads_collapsed_vs_genome.arf | sort | uniq | wc -l
588859
conda deactivate
Run mirdeep2. nano AST-2755_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2755
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2 on sample AST-2755 trimmed to 30bp" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/17_sed.collapse.cat.AST-2755.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/AST-2755/AST-2755_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2 concluded for sample AST-2755 trimmed to 30bp" $(date)
conda deactivate
Submitted batch job 293789. All of the mirdeep2 samples finished running today, yay!!!
20240130
I am now looking at all of the output data in an R script and I am thinking that I should concatenate all of the reads together, regardless of timepoint or treatment. Since its so many files, I’m going to write a script to concatenate and collapse
In the scripts folder: nano cat_collapse_all.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
echo "Concatenate and collapse smRNA reads from ALL samples" $(date)
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
# Concatenate reads
cat AST*.fastq > cat.all.fastq
echo "Reads concatenated, start collapse" $(date)
# Collapse concatenated reads
fastx_collapser -v -i cat.all.fastq -o collapse.cat.all.fastq
echo "Reads collapsed, start header adjustments for mirdeep2" $(date)
sed '/^>/ s/-/_x/g' collapse.cat.all.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.all.fastq
echo "Headers adjusted, start removing sequences <17 nts" $(date)
# Define the input and output files
input_file="sed.collapse.cat.all.fastq"
output_file="17_sed.collapse.cat.all.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.all.fastq
echo "Sequences removed, ready for mirdeep2" $(date)
Submitted batch job 293863. Ran into this error: fastx_collapser: Error: invalid quality score data on line 234199948 (quality_tok = "AAFFFJAJJJJJJJJJJJJJJJJJJJJ@GWNJ-0957:1001:GW2306054826th:4:1101:1560:1801 1:N:0:GTAGAGAT". Maybe the concatenate step didn’t work so well? I’m not sure why it would have failed but maybe the samples ‘catted’ together so that the first line of one sample ended up with the last line of another sample.
Trying to look at the line where things errored out:
sed -n '234199948,+20p' cat.all.fastq
AAFFFJAJJJJJJJJJJJJJJJJJJJJ
@GWNJ-0957:1001:GW2306054826th:4:1101:1560:1801 1:N:0:GTAGAGAT
CACCCCTCTTCCAATAACTTTACCTCTTA
+
AAAFA<F<FFJFJFA-FFJF-<FFJJJF7
@GWNJ-0957:1001:GW2306054826th:4:1101:1621:1801 1:N:0:GTAGAGAT
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
+
AAAFFJJJJJA--FJ7AAJFAJ-FF<F-7J
@GWNJ-0957:1001:GW2306054826th:4:1101:2067:1801 1:N:0:GTAGAGAT
TTTTGAAATCTAGAAGCTATGAAACT
+
AAAFFJJJJJJJFFJ<FFFF<JJJJJ
@GWNJ-0957:1001:GW2306054826th:4:1101:2087:1801 1:N:0:GTAGAGAT
TGATCTTGTAGGTTCCATCTTAT
+
AA<AA<FFJ7F7<7AAAJFJFJF
@GWNJ-0957:1001:GW2306054826th:4:1101:2189:1801 1:N:0:GTAGAGAT
GCCTTTGTGCTATGATCTGTTGAGGTTCTG
+
AA<AFJJJFJJJ7AFFJJJFFJJFJJJ-F-
@GWNJ-0957:1001:GW2306054826th:4:1101:3224:1801 1:N:0:GTAGAGAT
I appear to be correct in that one line was catted to the end of another line. How to prevent this???? Idk if this is even the only instance where this happened in the cat file. I added the line below to the cat_collapse_all.sh script after the reads are concatenated and before the collapsing.
sed 's/@/\n@/g' cat.all.fastq > check.cat.all.fastq
Submitted batch job 293874. Now I’m getting this error: fastx_collapser: input file (check.cat.all.fastq) has unknown file format (not FASTA or FASTQ), first character = (10). I’m adding this line in after the sed line: grep -v '^[[:space:]]*$' check.cat.all.fastq > check.cat.all.fastq. This should remove any blank lines in the data. Submitted batch job 293876
20240130
Now I’m getting this error: grep: input file ‘check.cat.all.fastq’ is also the output. fastx_collapser: Premature End-Of-File (filename ='check.cat.all.fastq'). I’m going to rename the grep output so that it is grep.check.cat.all.fastq and change the input file name to grep.check.cat.all.fastq for fastx. Submitted batch job 293889. Ran for 3 hours and then failed. This is the error: fastx_collapser: Error: invalid quality score data on line 234199948 (quality_tok = "AAFFFJAJJJJJJJJJJJJJJJJJJJJ". AGGGGGGGHHHHHHGGHHHH. I don’t know what to do…Okay Chat gpt wrote an awk script for me. In the scripts folder, I did nano filter_length.awk and put the following code in:
awk '{
# Read the header line
header = $0
# Read the sequence line
getline
sequence = $0
# Read the "+" line
getline
# Read the quality score line
quality = $0
# Check if sequence length matches quality score length
if (length(sequence) == length(quality)) {
# Print the record
print header
print sequence
print "+"
print quality
}
}' input.fastq > output.fastq
This code SHOULD remove any sequence whose length differs from the length of its quality score lines. I added this line: awk -f /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/filter_length.awk grep.check.cat.all.fastq > awk.grep.check.cat.all.fastq below the grep line. Now cat_collapse_all.sh looks like this:
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
echo "Concatenate and collapse smRNA reads from ALL samples" $(date)
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
# Concatenate reads
#cat AST*.fastq > cat.all.fastq
# Make sure there are no issues with cat - ensure that every @ symbol is a new line
echo "Make sure all lines start with @ symbol" $(date)
sed 's/@/\n@/g' cat.all.fastq > check.cat.all.fastq
echo "Remove any blank lines" $(date)
grep -v '^[[:space:]]*$' check.cat.all.fastq > grep.check.cat.all.fastq
echo "Remove any sequences where the length of the sequence and length of quality score differ" $(date)
awk -f /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/filter_length.awk grep.check.cat.all.fastq > awk.grep.check.cat.all.fastq
echo "Reads concatenated, start collapse" $(date)
# Collapse concatenated reads
fastx_collapser -v -i awk.grep.check.cat.all.fastq -o collapse.cat.all.fastq
echo "Reads collapsed, start header adjustments for mirdeep2" $(date)
sed '/^>/ s/-/_x/g' collapse.cat.all.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.all.fastq
echo "Headers adjusted, start removing sequences <17 nts" $(date)
# Define the input and output files
input_file="sed.collapse.cat.all.fastq"
output_file="17_sed.collapse.cat.all.fastq"
# Initialize the output file
> "$output_file"
# Use awk to process the sequences
awk '{
if (substr($0, 1, 1) == ">") {
header = $0
getline
sequence = $0
if (length(sequence) >= 17) {
print header >> "'$output_file'"
print sequence >> "'$output_file'"
}
}
}' "$input_file"
zgrep -c ">" 17_sed.collapse.cat.all.fastq
echo "Sequences removed, ready for mirdeep2" $(date)
Submitted batch job 293969
20240201
NOW I got this error:
awk: /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/filter_length.awk:1: awk '{
awk: /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/filter_length.awk:1: ^ invalid char ''' in expression
awk: /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/filter_length.awk:1: awk '{
awk: /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/filter_length.awk:1: ^ syntax error
fastx_collapser: Premature End-Of-File (filename ='awk.grep.check.cat.all.fastq')
I might just manually do it. I also rezipped all of the files in my flexbar trimmed data folder.
20240202
I am going to follow the concatenate step in Sam’s code. Here’s his code. He does a lot of echoing and what not.
# Load bash variables into memory
source .bashvars
# Check for existence of concatenated FastA before running
if [ ! -f "${output_dir_top}/${concatenated_trimmed_reads_fastq}" ]; then
cat ${trimmed_fastqs_dir}/*.fastq.gz \
> "${output_dir_top}/${concatenated_trimmed_reads_fastq}"
fi
ls -lh "${output_dir_top}/${concatenated_trimmed_reads_fastq}"
Let’s try to modify it for my info. First, I’m just going to concatenate. In the scripts folder: nano cat_all.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load FASTX-Toolkit/0.0.14-GCC-9.3.0
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
echo "Concatenate smRNA reads from ALL samples" $(date)
# Check for existence of concatenated FastA before running
if [ ! -f "cat.all.fastq" ]; then
cat AST*.fastq.gz \
> cat.all.fastq
fi
echo "Reads concatenated" $(date)
Submitted batch job 294079. Ran in about 2 mins but produced a binary file which is weird…Going to edit the code so that it is just cat AST*.fastq.gz. Submitted batch job 294080. Took about 2 mins. Once again, produced a binary file. Maybe because I am concatenating .gz files? Let me try to run fastx_collapse and see if it works. In the scripts folder: nano collapse_all.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
echo "Collapse concatenated reads" $(date)
fastx_collapser -v -i cat.all.fastq -o collapse.cat.all.fastq
echo "Reads collapsed" $(date)
Submitted batch job 294081. Failed immediately and got this error: fastx_collapser: input file (cat.all.fastq) has unknown file format (not FASTA or FASTQ), first character = ^_ (31). Okay I might go back to basics…so the cat step worked when I was just catting R1 and R2 together. Maybe lets try to do that with two samples and then cat those samples together and see if they collapse.
interactive
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
gunzip AST-1065_R1_001.fastq*
gunzip AST-1105_R1_001.fastq*
cat AST-1065_R1_001.fastq.gz_1.fastq AST-1065_R1_001.fastq.gz_2.fastq > AST-1065.fastq
cat AST-1105_R1_001.fastq.gz_1.fastq AST-1105_R1_001.fastq.gz_2.fastq > AST-1105.fastq
cat AST-1065.fastq AST-1105.fastq > test.fastq
module load FASTX-Toolkit/0.0.14-GCC-9.3.0
fastx_collapser -v -i AST-1065.fastq -o collapse.AST-1065.fastq
Input: 35658222 sequences (representing 35658222 reads)
Output: 11979585 sequences (representing 35658222 reads)
Okay so fastx is happy with the R1 and R2 concatenated output. Now lets try on the test.fastq file.
fastx_collapser -v -i test.fastq -o collapse.test.fastq
Got this error: fastx_collapser: Error: invalid quality score data on line 234199948 (quality_tok = "AAFFFJAJJJJJJJJJJJJJJJJJJJJ". Let’s look at the line where the error is occurring. Going to set it 10 lines up so that I can see whats going on
sed -n '234199948, 234199948p' test.fastq
AAFFFJAJJJJJJJJJJJJJJJJJJJJ
Does it hate the J ascii character???
I am now realizing that AST-1105 was one of the samples that looked weird and that I excluded from analysis. Let me choose a different sample to test with AST-1065
gunzip AST-2404_R1_001.fastq.gz*
cat AST-2404_R1_001.fastq.gz_1.fastq AST-2404_R1_001.fastq.gz_2.fastq > AST-2404.fastq
Let’s look at how many lines are in each file
wc -l AST-1065.fastq
142632888 AST-1065.fastq
wc -l AST-2404.fastq
133703224 AST-2404.fastq
Since I am catting these two files, 142632888 lines + 133703224 lines = 276336112 lines – this is how many lines should be in the fastq file once I cat them together.
cat AST-1065.fastq AST-2404.fastq > test.fastq
wc -l test.fastq
276336112 test.fastq
Expected number of lines. Let’s try to collapse
fastx_collapser -v -i test.fastq -o collapse.test.fastq
Input: 69084028 sequences (representing 69084028 reads)
Output: 22118660 sequences (representing 69084028 reads)
Took a while but worked!!! Maybe it is the problem sample AST-1105 that is messing everything up…going to move it to its own folder in the flexbar folder and rerun the script.
mkdir AST-1105
mv *1105* AST-1105
cd ../../../scripts/
sbatch cat_all.sh
Submitted batch job 294086. Took 2 mins but still a binary file…Do I have to just do it manually??? Maybe instead I will cat all of the R1 and R2 reads together and then cat those files together.
We had the e5 molecular meeting and Sam White said that Azenta recommended that we toss read 2 and don’t use it at all. So I may try to just cat the R1…I’m going to edit the cat_all.sh script. Now here’s what it says:
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load FASTX-Toolkit/0.0.14-GCC-9.3.0
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
echo "Unzip R1 files" $(date)
gunzip AST*_1.fastq.gz
echo "Unzipping complete, concatenate smRNA reads from ALL samples - R1 only" $(date)
cat AST*_1.fastq > cat.all.fastq
echo "R1 concatenated" $(date)
Submitted batch job 294088. Took about 5 mins. Looks good!! Finally successful cat (so far). Now running the collapse_all.sh script. Submitted batch job 294089
20240204
Success!!!!!!! Reads have been concatenated!!!!! At least the R1 reads. Here’s the output message:
Collapse concatenated reads Fri Feb 2 14:31:47 EST 2024
Input: 343437674 sequences (representing 343437674 reads)
Output: 62419423 sequences (representing 343437674 reads)
Reads collapsed Fri Feb 2 16:28:11 EST 2024
Go to the file in /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar and check it out
head collapse.cat.all.fastq
>1-8268409
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>2-2708289
GCACTGGTGGTTCAGTGGTAGAATTCTC
>3-2338875
GACTTTGTAGCATAGGTAAGGTTAGTGCAT
>4-2210232
AACTTTTGACGGTGGATCTCTTGGCTCACG
>5-1864084
TAAGACTATGATTATATGCAGCTTCTTGCA
tail collapse.cat.all.fastq
>62419419-1
ATTTGACGAAGGCTCCAAAGGAAGTCATGG
>62419420-1
TTGCCCGTATTACTGCCGT
>62419421-1
TACCTGCCCTATTTGCCTTATACTAG
>62419422-1
CTGGAAATCTGCTGGACTTACGTTT
>62419423-1
CGACAGTGGGCTGAAGCTG
zgrep -c ">" collapse.cat.all.fastq
62419423
Prep the sequence headers for mirdeep2 analysis
sed '/^>/ s/-/_x/g' collapse.cat.all.fastq \
| sed '/^>/ s/>/>seq_/' \
> sed.collapse.cat.all.fastq
head sed.collapse.cat.all.fastq
>seq_1_x8268409
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_2_x2708289
GCACTGGTGGTTCAGTGGTAGAATTCTC
>seq_3_x2338875
GACTTTGTAGCATAGGTAAGGTTAGTGCAT
>seq_4_x2210232
AACTTTTGACGGTGGATCTCTTGGCTCACG
>seq_5_x1864084
TAAGACTATGATTATATGCAGCTTCTTGCA
zgrep -c ">" sed.collapse.cat.all.fastq
62419423
Now I can run mirdeep2 (mapping and prediction steps). Because the input fasta file will likely be so big, I’m going to include the mapper.pl with the mirdeep2.pl script. First, make a new folder in the mirdeep2 folder for all samples.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2
mkdir all
Run mirdeep2. In the scripts folder: nano mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mapping" $(date)
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/sed.collapse.cat.all.fastq -c -p /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts/Apoc_ref.btindex -s /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/all_reads_collapsed.fa -t /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/all_reads_collapsed_vs_genome.arf -v
echo "Mapping complete, Starting mirdeep2" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/sed.collapse.cat.all.fastq /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/all_reads_collapsed_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -t N.vectensis -P -v -g -1 2>report.log
echo "mirdeep2" $(date)
conda deactivate
Submitted batch job 294127
20240206
Success! mirdeep2 finished running, took about 1.5 days. I’m going to download the results to my computer. Since the output is in the scripts folder, I’m going to move all of the output to the all folder. Here’s a summary of the output:
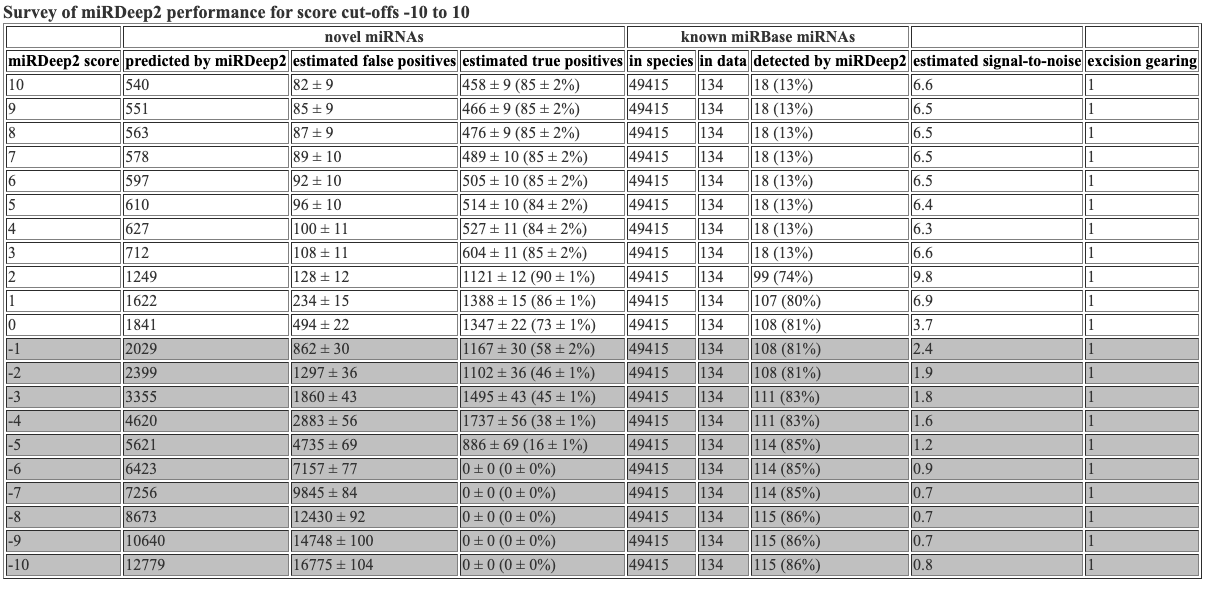
I think my next steps will be filtering in R. I’m going to filter the csv so that I retain potential miRNAs that have an mirdeep2 score > 10, no rfam info, at least 10 reads in mature and star read count, and significant randfold pvalue (this has been done in most of the other cnidarian miRNA papers). When I did this filtering in this script, I ended up with 278 novel miRNAs.
Additionally, I will need to write a script that looks at: ““requirement of a 2-nucleotide overhang on the 3’ end of the precursor miRNA, 5’ consistency of the mature miRNA strand (at least 90% of the reads have to be starting from the same position), and at least 16 nucleotide complementarity between mature and star strand” (Praher et al., 2021). I may do this manually by looking at the PDFs…
I also need to figure out how MFE is calculated?
After I do that, I will probably blast the predicted seqs against a tRNA and rRNA database to remove any unwanted RNAs. RNAcentral gives a good overview of the different ncRNA databases that could be used. Here are some options:
- rRNA
- tRNA
I should also blast it against the NCBI database.
20240226
Since mirdeep2 has run with all R1 reads concatenated, I can now run the quantifier module for each sample. I think I might need to collapse each R1 file and modify it with the correct headers. From the mirdeep2 github, it says the input for the quantifier module is:
- A FASTA file with precursor sequences,
- a FASTA file with mature miRNA sequences,
- a FASTA file with deep sequencing reads, and
- optionally a FASTA file with star sequences and the 3 letter code of the species of interest.
The fasta files with the precursor, mature and star sequences are in this folder: /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57. The fasta files are separated by known and novel, so I will cat the known and novel sequences together.
cat known_mature_04_02_2024_t_11_15_57_score-50_to_na.fa novel_mature_04_02_2024_t_11_15_57_score-50_to_na.fa > mature_all.fa
cat known_pres_04_02_2024_t_11_15_57_score-50_to_na.fa novel_pres_04_02_2024_t_11_15_57_score-50_to_na.fa > precursor_all.fa
cat known_star_04_02_2024_t_11_15_57_score-50_to_na.fa novel_star_04_02_2024_t_11_15_57_score-50_to_na.fa > star_all.fa
Let’s try to run a sample without collapsing it.
conda activate /data/putnamlab/mirdeep2
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -s /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/star_all.fa -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq
As I suspected, it gave me this error:
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq ids do not have the correct format
it must have the id line >SSS_INT_xINT
SSS is a three letter code indicating the sample origin
INT is just a running number
xINT is the number of read occurrences
But it did give me this recommendation:
You can use the mapper.pl module to create such a file from a fasta file with
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq.collapsed
So lets give that a try! Ran in about a minute. Let’s try to run the quantifier module now.
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -s /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/star_all.fa -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq.collapsed
Ran in about a minute and gave this output:
getting samples and corresponding read numbers
Converting input files
building bowtie index
mapping mature sequences against index
mapping read sequences against index
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 17829111 43551 17785560 0.244 99.756
seq: 17829111 43551 17785560 0.244 99.756
mapping star sequences against index
analyzing data
1873 mature mappings to precursors
1865 star mappings to precursors
Expressed miRNAs are written to expression_analyses/expression_analyses_1708999755/miRNA_expressed.csv
not expressed miRNAs are written to expression_analyses/expression_analyses_1708999755/miRNA_not_expressed.csv
Creating miRBase.mrd file
Mapped READS readin - DONE
make_html2.pl -q expression_analyses/expression_analyses_1708999755/miRBase.mrd -k mature_all.fa -y 1708999755 -o -i expression_analyses/expression_analyses_1708999755/mature_all.fa_mapped.arf -j expression_analyses/expression_analyses_1708999755/star_all.fa_mapped.arf -l -M miRNAs_expressed_all_samples_1708999755.csv
miRNAs_expressed_all_samples_1708999755.csv file with miRNA expression values
parsing miRBase.mrd file finished
creating PDF files
Can't use string ("29") as a HASH ref while "strict refs" in use at /data/putnamlab/mirdeep2/bin/make_html2.pl line 658.
I also ran the code so that the -s argument was removed. It gave the same mapping but it did print out creating pdf for chromosome_XXXX while the other line of code didn’t. I looked at less miRNAs_expressed_all_samples_1708999755.csv
#miRNA read_count precursor total seq seq(norm)
chromosome_10_365643 2.00 chromosome_10_365643 2.00 2.00 33.80
chromosome_10_365701 2.00 chromosome_10_365701 2.00 2.00 33.80
chromosome_10_366823 1.00 chromosome_10_366823 1.00 1.00 16.90
chromosome_10_366897 229.00 chromosome_10_366897 229.00 229.00 3870.47
chromosome_10_367039 2.00 chromosome_10_367039 2.00 2.00 33.80
chromosome_10_367612 0.00 chromosome_10_367612 0.00 0 0
chromosome_10_367894 56.00 chromosome_10_367894 56.00 56.00 946.49
chromosome_10_368110 0.00 chromosome_10_368110 0.00 0 0
chromosome_10_368443 0.00 chromosome_10_368443 0.00 0 0
chromosome_10_370371 11.00 chromosome_10_370371 11.00 11.00 185.92
chromosome_10_370480 0.00 chromosome_10_370480 0.00 0 0
chromosome_10_370856 2.00 chromosome_10_370856 2.00 2.00 33.80
I’m not sure what read count, seq and seq(norm) means…but this file looks different than miRNA_expressed.csv, which looks like:
#miRNA read_count precursor
chromosome_10_365643 2 chromosome_10_365643
chromosome_10_365701 2 chromosome_10_365701
chromosome_10_366823 1 chromosome_10_366823
chromosome_10_366897 229 chromosome_10_366897
chromosome_10_367039 2 chromosome_10_367039
chromosome_10_367612 0 chromosome_10_367612
chromosome_10_367894 56 chromosome_10_367894
chromosome_10_368110 0 chromosome_10_368110
chromosome_10_368443 0 chromosome_10_368443
chromosome_10_370371 11 chromosome_10_370371
chromosome_10_370480 0 chromosome_10_370480
chromosome_10_370856 2 chromosome_10_370856
chromosome_10_371859 1 chromosome_10_371859
chromosome_10_371901 0 chromosome_10_371901
It looks like they have the same read counts but what does seq(norm) mean???? I need to read this page. I think I would just use the read count info but the counts seem so low. This is also just one sample and I’m not seeing all of the potential miRNAs when I just look at the data briefly.
Also maybe look at this: https://www.biorxiv.org/content/10.1101/2021.10.19.464446v1.full.pdf
It would be interesting to try the -W and -k tags in the quantifier module. -W indicates that read counts are weighed by their number of mappings. e.g. A read maps twice so each position gets 0.5 added to its read profile. -k also considers precursor-mature mappings that have different ids, eg let7c would be allowed to map to pre-let7a.
20240227
Going to continue looking at the mirdeep2 quantifier output. When looking at other miRNA cnidarian papers (eg Liew et al. 2014, Baumgarten et al.), it seems like my counts (at least for this one sample) are comparable. Let’s try with the -W tag.
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -W -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq.collapsed
getting samples and corresponding read numbers
Converting input files
building bowtie index
mapping mature sequences against index
mapping read sequences against index
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 17829111 43551 17785560 0.244 99.756
seq: 17829111 43551 17785560 0.244 99.756
analyzing data
1873 mature mappings to precursors
Expressed miRNAs are written to expression_analyses/expression_analyses_1709042138/miRNA_expressed.csv
not expressed miRNAs are written to expression_analyses/expression_analyses_1709042138/miRNA_not_expressed.csv
Creating miRBase.mrd file
Mapped READS readin - DONE
make_html2.pl -q expression_analyses/expression_analyses_1709042138/miRBase.mrd -k mature_all.fa -y 1709042138 -o -i expression_analyses/expression_analyses_1709042138/mature_all.fa_mapped.arf -l -M miRNAs_expressed_all_samples_1709042138.csv -W expression_analyses/expression_analyses_1709042138/read_occ
miRNAs_expressed_all_samples_1709042138.csv file with miRNA expression values
parsing miRBase.mrd file finished
Took a few mins to run and generate the pdfs. Let’s look at the expressed file:
#miRNA read_count precursor
chromosome_10_365643 2 chromosome_10_365643
chromosome_10_365701 2 chromosome_10_365701
chromosome_10_366823 0.5 chromosome_10_366823
chromosome_10_366897 229 chromosome_10_366897
chromosome_10_367039 2 chromosome_10_367039
chromosome_10_367612 0 chromosome_10_367612
chromosome_10_367894 3.83455475552999 chromosome_10_367894
chromosome_10_368110 0 chromosome_10_368110
chromosome_10_368443 0 chromosome_10_368443
chromosome_10_370371 0.703267973856209 chromosome_10_370371
chromosome_10_370480 0 chromosome_10_370480
chromosome_10_370856 2 chromosome_10_370856
chromosome_10_371859 1 chromosome_10_371859
chromosome_10_371901 0 chromosome_10_371901
Interesting. So with the -W tag, read counts are weighed by their number of mappings. e.g. A read maps twice so each position gets 0.5 added to its read profile. So for instance, does that mean that chromosome_10_366823 has 2 reads mapped to it? That’s confusing.
Let’s try with the -k tag.
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -k -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq.collapsed
getting samples and corresponding read numbers
Converting input files
building bowtie index
mapping mature sequences against index
mapping read sequences against index
Mapping statistics
#desc total mapped unmapped %mapped %unmapped
total: 17829111 43551 17785560 0.244 99.756
seq: 17829111 43551 17785560 0.244 99.756
analyzing data
6029 mature mappings to precursors
Expressed miRNAs are written to expression_analyses/expression_analyses_1709043340/miRNA_expressed.csv
not expressed miRNAs are written to expression_analyses/expression_analyses_1709043340/miRNA_not_expressed.csv
Creating miRBase.mrd file
Mapped READS readin - DONE
make_html2.pl -q expression_analyses/expression_analyses_1709043340/miRBase.mrd -k mature_all.fa -y 1709043340 -o -i expression_analyses/expression_analyses_1709043340/mature_all.fa_mapped.arf -M miRNAs_expressed_all_samples_1709043340.csv
miRNAs_expressed_all_samples_1709043340.csv file with miRNA expression values
parsing miRBase.mrd file finished
Not sure what is different looking at the output files. It is giving me more mature mappings to precursors because I set it so that it would map to multiple locations.
I think I am just going to stick with the original code without the star sequence information. Going to now process the rest of the samples. I moved all of the stuff I did above into a folder called test (/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/test). First, going to run the mapper module to collapse the samples. I could definitely do this in a script but I want to do it manually the first time so that I am understanding how the code works.
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq.collapsed
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1147_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1147_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_156798
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1412_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1412_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157142
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1560_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1560_R1_001.fastq.gz_1.fastq.collapsed
Log file for this run is in mapper_logs and called mapper.log_187756
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1567_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1567_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157352
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1617_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1617_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157469
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1722_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1722_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157568
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2000_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2000_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157668
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2007_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2007_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157831
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2302_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2302_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_157928
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2360_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2360_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158073
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2398_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2398_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158172
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2404_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2404_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158270
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2412_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2412_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158370
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2512_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2512_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158493
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2523_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2523_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158577
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2563_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2563_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158695
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2729_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2729_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158793
mapper.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2755_R1_001.fastq.gz_1.fastq -e -m -h -s /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2755_R1_001.fastq.gz_1.fastq.collapsed
# Log file for this run is in mapper_logs and called mapper.log_158919
This is what the top lines of one of the collapsed files looks like:
head AST-2755_R1_001.fastq.gz_1.fastq.collapsed
>seq_0_x282696
GCACTGGTGGTTCAGTGGTAGAATTCTCGC
>seq_282696_x73794
TGAAAATCTTTTCTCTGAAGTGGAA
>seq_356490_x65651
TGACTAGATATATACTCATGCT
>seq_422141_x62171
GCACTGATGGTTCAGTGGTAGAATTCTCGC
>seq_484312_x61289
TGACTAGATATACACTCATTCT
Count the number of unique sequences in each collapsed file.
zgrep -c ">" *fastq.collapsed
AST-1065_R1_001.fastq.gz_1.fastq.collapsed:5255291
AST-1147_R1_001.fastq.gz_1.fastq.collapsed:7375460
AST-1412_R1_001.fastq.gz_1.fastq.collapsed:4942609
AST-1567_R1_001.fastq.gz_1.fastq.collapsed:4255807
AST-1617_R1_001.fastq.gz_1.fastq.collapsed:3410530
AST-1560_R1_001.fastq.gz_1.fastq.collapsed:5054135
AST-1722_R1_001.fastq.gz_1.fastq.collapsed:3144650
AST-2000_R1_001.fastq.gz_1.fastq.collapsed:4692561
AST-2007_R1_001.fastq.gz_1.fastq.collapsed:3658938
AST-2302_R1_001.fastq.gz_1.fastq.collapsed:4891546
AST-2360_R1_001.fastq.gz_1.fastq.collapsed:4165438
AST-2398_R1_001.fastq.gz_1.fastq.collapsed:4065976
AST-2404_R1_001.fastq.gz_1.fastq.collapsed:5091971
AST-2412_R1_001.fastq.gz_1.fastq.collapsed:4462387
AST-2512_R1_001.fastq.gz_1.fastq.collapsed:4235505
AST-2523_R1_001.fastq.gz_1.fastq.collapsed:5171942
AST-2563_R1_001.fastq.gz_1.fastq.collapsed:5147628
AST-2729_R1_001.fastq.gz_1.fastq.collapsed:4622195
AST-2755_R1_001.fastq.gz_1.fastq.collapsed:4816214
Retained 3-7 million unique reads per sample. Run quantifier module on all samples. Again, I could definitely do this in a script but I want to do it manually the first time so that I am understanding how the code works.
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 17829111 43551 17785560 0.244 99.756
#seq: 17829111 43551 17785560 0.244 99.756
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709048527/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1147_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 40415224 869326 39545898 2.151 97.849
#seq: 40415224 869326 39545898 2.151 97.849
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709052026/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1412_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16279555 69729 16209826 0.428 99.572
#seq: 16279555 69729 16209826 0.428 99.572
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709053021/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1560_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 17827024 6510 17820514 0.037 99.963
#seq: 17827024 6510 17820514 0.037 99.963
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709054526/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1567_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16611397 105945 16505452 0.638 99.362
#seq: 16611397 105945 16505452 0.638 99.362
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709053231/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1617_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16077717 121046 15956671 0.753 99.247
#seq: 16077717 121046 15956671 0.753 99.247
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055134/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1722_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16430221 258350 16171871 1.572 98.428
#seq: 16430221 258350 16171871 1.572 98.428
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055319/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2000_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 17428854 91821 17337033 0.527 99.473
#seq: 17428854 91821 17337033 0.527 99.473
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055397/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2007_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16559551 102426 16457125 0.619 99.381
#seq: 16559551 102426 16457125 0.619 99.381
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055480/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2302_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16665370 230870 16434500 1.385 98.615
#seq: 16665370 230870 16434500 1.385 98.615
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055687/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2360_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16648356 20477 16627879 0.123 99.877
#seq: 16648356 20477 16627879 0.123 99.877
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055924/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2398_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16788208 230412 16557796 1.372 98.628
#seq: 16788208 230412 16557796 1.372 98.628
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709055986/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2404_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16712903 128490 16584413 0.769 99.231
#seq: 16712903 128490 16584413 0.769 99.231
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057037/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2412_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 17488508 27520 17460988 0.157 99.843
#seq: 17488508 27520 17460988 0.157 99.843
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057124/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2512_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16265716 14678 16251038 0.090 99.910
#seq: 16265716 14678 16251038 0.090 99.910
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057192/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2523_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16995265 88993 16906272 0.524 99.476
#seq: 16995265 88993 16906272 0.524 99.476
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057251/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2563_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 17023002 70635 16952367 0.415 99.585
#seq: 17023002 70635 16952367 0.415 99.585
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057330/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2729_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 17121869 23317 17098552 0.136 99.864
#seq: 17121869 23317 17098552 0.136 99.864
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057412/miRNA_expressed.csv
quantifier.pl -p /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/precursor_all.fa -m /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -d -r /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2755_R1_001.fastq.gz_1.fastq.collapsed
#desc total mapped unmapped %mapped %unmapped
#total: 16269823 127434 16142389 0.783 99.217
#seq: 16269823 127434 16142389 0.783 99.217
#analyzing data
#1873 mature mappings to precursors
#Expressed miRNAs are written to expression_analyses/expression_analyses_1709057479/miRNA_expressed.csv
Mapped % are very low for all samples (<1%). This does not surprise me since miRNAs aren’t super abundant in the genome. For each sample, a miRNAs_expressed_all_samples_XXXXX.csv file got produced, as well as files of miRNAs expressed and not expressed. Look at how many lines are in the miRNA expressed all samples file for each sample:
wc -l miRNAs_expressed_all_samples*
1874 miRNAs_expressed_all_samples_1709048527.csv
1874 miRNAs_expressed_all_samples_1709052026.csv
1874 miRNAs_expressed_all_samples_1709052927.csv
1874 miRNAs_expressed_all_samples_1709053021.csv
1874 miRNAs_expressed_all_samples_1709053231.csv
1874 miRNAs_expressed_all_samples_1709054272.csv
1874 miRNAs_expressed_all_samples_1709054526.csv
1874 miRNAs_expressed_all_samples_1709055134.csv
1874 miRNAs_expressed_all_samples_1709055319.csv
1874 miRNAs_expressed_all_samples_1709055397.csv
1874 miRNAs_expressed_all_samples_1709055480.csv
1874 miRNAs_expressed_all_samples_1709055687.csv
1874 miRNAs_expressed_all_samples_1709055924.csv
1874 miRNAs_expressed_all_samples_1709055986.csv
1874 miRNAs_expressed_all_samples_1709057037.csv
1874 miRNAs_expressed_all_samples_1709057124.csv
1874 miRNAs_expressed_all_samples_1709057192.csv
1874 miRNAs_expressed_all_samples_1709057251.csv
1874 miRNAs_expressed_all_samples_1709057330.csv
1874 miRNAs_expressed_all_samples_1709057412.csv
1874 miRNAs_expressed_all_samples_1709057479.csv
39354 total
All have the same number of lines. Are they in the same order?
# Top of file
head miRNAs_expressed_all_samples_1709048527.csv
#miRNA read_count precursor total seq seq(norm)
chromosome_10_365643 2.00 chromosome_10_365643 2.00 2.00 36.63
chromosome_10_365701 2.00 chromosome_10_365701 2.00 2.00 36.63
chromosome_10_366823 1.00 chromosome_10_366823 1.00 1.00 18.31
chromosome_10_366897 229.00 chromosome_10_366897 229.00 229.00 4193.60
chromosome_10_367039 2.00 chromosome_10_367039 2.00 2.00 36.63
chromosome_10_367612 0.00 chromosome_10_367612 0.00 0 0
chromosome_10_367894 56.00 chromosome_10_367894 56.00 56.00 1025.51
chromosome_10_368110 0.00 chromosome_10_368110 0.00 0 0
chromosome_10_368443 0.00 chromosome_10_368443 0.00 0 0
head miRNAs_expressed_all_samples_1709057479.csv
#miRNA read_count precursor total seq seq(norm)
chromosome_10_365643 10.00 chromosome_10_365643 10.00 10.00 58.13
chromosome_10_365701 0.00 chromosome_10_365701 0.00 0 0
chromosome_10_366823 10.00 chromosome_10_366823 10.00 10.00 58.13
chromosome_10_366897 1.00 chromosome_10_366897 1.00 1.00 5.81
chromosome_10_367039 8.00 chromosome_10_367039 8.00 8.00 46.50
chromosome_10_367612 0.00 chromosome_10_367612 0.00 0 0
chromosome_10_367894 54.00 chromosome_10_367894 54.00 54.00 313.89
chromosome_10_368110 0.00 chromosome_10_368110 0.00 0 0
chromosome_10_368443 15.00 chromosome_10_368443 15.00 15.00 87.19
# Bottom of file
tail miRNAs_expressed_all_samples_1709048527.csv
chromosome_9_363461 0.00 chromosome_9_363461 0.00 0 0
chromosome_9_363522 3.00 chromosome_9_363522 3.00 3.00 54.94
chromosome_9_363632 0.00 chromosome_9_363632 0.00 0 0
chromosome_9_363719 6.00 chromosome_9_363719 6.00 6.00 109.88
chromosome_9_364029 3.00 chromosome_9_364029 3.00 3.00 54.94
chromosome_9_364032 0.00 chromosome_9_364032 0.00 0 0
chromosome_9_364034 49.00 chromosome_9_364034 49.00 49.00 897.32
chromosome_9_364498 0.00 chromosome_9_364498 0.00 0 0
chromosome_9_364714 13.00 chromosome_9_364714 13.00 13.00 238.06
chromosome_9_364995 2.00 chromosome_9_364995 2.00 2.00 36.63
tail miRNAs_expressed_all_samples_1709057479.csv
chromosome_9_363461 6.00 chromosome_9_363461 6.00 6.00 34.88
chromosome_9_363522 4.00 chromosome_9_363522 4.00 4.00 23.25
chromosome_9_363632 0.00 chromosome_9_363632 0.00 0 0
chromosome_9_363719 5.00 chromosome_9_363719 5.00 5.00 29.06
chromosome_9_364029 33.00 chromosome_9_364029 33.00 33.00 191.82
chromosome_9_364032 0.00 chromosome_9_364032 0.00 0 0
chromosome_9_364034 159.00 chromosome_9_364034 159.00 159.00 924.24
chromosome_9_364498 2.00 chromosome_9_364498 2.00 2.00 11.63
chromosome_9_364714 9.00 chromosome_9_364714 9.00 9.00 52.32
chromosome_9_364995 7.00 chromosome_9_364995 7.00 7.00 40.69
From a quick look at the top and bottom of files, it looks like they are in the same order. I copied all miRNAs_expressed_all_samples_XXXX.csv files to my local computer and renamed each file so that the sample ID was in the name of the file. Now I’ll look at them in R.
20240305
Still trying to figure out how the heck MFE is calculated and where that number is stored in the files that mirdeep2 produces. I looked through all of the files and STILL CAN’T FIND IT…
I do think that the files mature_vs_precursors.bwt and reads_vs_precursors.bwt in /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/dir_prepare_signature1707063877 will help me calculate if 90% of the reads share the same nucleotide start at the 5’ end.
20240319
I have now identified all putative miRNAs (code here) and run DESeq2 (code here). For both the mRNAs and the miRNAs, I now have a list of unique differentially expressed genes or miRNAs. I can use these lists to filter the gene and miRNA fastas so that I can run miranda with the subsetted sequences.
I need to do mirnda with the 3’ UTR of the mRNA, meaning I have to use the gff to identify the 3’ UTR in the genome and then subset those sequences specifically…When looking at the Astrangia gff, there are only 687 3’ UTR rows in the gff, despite there being >48,000 genes. Somehow, I need to get the sequences of the 3 UTRs for all mRNA sequences.
These are some options for IDing the 3’ end:
- https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-4241-1
- GETUTR
- 3USS
- UTRscan
- Maker
- Augustus
I know this won’t be the same, but I am going to run blast with the query as the miRNA sequences and the reference db as the mRNA sequences. In the scripts folder: nano blastn_miRNA.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
echo "Making blast db from mRNA sequences" $(date)
makeblastdb -in /data/putnamlab/jillashey/Astrangia_Genome/apoculata_mrna_v2.0.fasta -dbtype 'nucl' -out /data/putnamlab/jillashey/Astrangia2021/smRNA/data/apoc_mRNA_db
echo "Blast db creation complete, blasting miRNAs against db" $(date)
blastn -query /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -db /data/putnamlab/jillashey/Astrangia2021/smRNA/data/apoc_mRNA_db -outfmt 6 -evalue 4 -num_threads 15 -out /data/putnamlab/jillashey/Astrangia2021/smRNA/data/blastn_miRNA_query.tab
echo "Blast complete!" $(date)
Submitted batch job 309649. Ran fast, but output file was empty…
20240320
Going to try to rerun the script above but removing any extraneous flags in hopes that this will fix the problem. Submitted batch job 309660. Still empty but not getting any error…I find it hard to believe that there are no blast matches between the miRNAs and the mRNAs…though I guess I am looking for complementary sequences, not identical sequences. Is there a way to do this in blast? Yes there is! I just need to add -strand both to allow BLAST to search both the forward and reverse-complement strands of the database sequences. Submitted batch job 309661. Still ran but still output file is empty………….Truly don’t understand and I am quite frustrated. Maybe let’s try blastx? In the scripts folder: nano blastx_miRNA.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
echo "Making blast db from mRNA sequences" $(date)
makeblastdb -in /data/putnamlab/jillashey/Astrangia_Genome/apoculata_proteins_v2.0.fasta -dbtype prot -out /data/putnamlab/jillashey/Astrangia2021/smRNA/data/apoc_prot_db
echo "Blast db creation complete, blasting miRNAs against db" $(date)
blastx -query /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa -db /data/putnamlab/jillashey/Astrangia2021/smRNA/data/apoc_prot_db -out /data/putnamlab/jillashey/Astrangia2021/smRNA/data/blastx_miRNA_query.txt -outfmt 6
echo "Blast complete!" $(date)
Submitted batch job 309665. That worked! It was very fast. I wonder why it’s not working with the nucleotide sequences…Maybe there really are no hits? I just find that hard to believe but idk maybe.
20240321
Coming to the realization that I either have to locate the 3’ UTRs in the genome myself or run a gene prediction software (augustus/maker) to find the 3’ UTR coordinates. My worry is that I use augustus or maker and not all of the 3’UTRs for the mRNAs are identified.
The other idea was to look at the stop codon coordinates for each mRNA and assume that everything after the stop codon sequence is the 3’UTR. This will probably take me less time but still not sure how to do it. After searching around, the bedtools suite may help me. Someone on a github issue page suggested the following:
“I recommend you get a BED file with the position of the CDS (the annotation is always correct….). You can then use some Bedtools operations: bedtools merge the different exons of your Isoscm gtf files which form a same gene (they are next to each other). Use bedtools substract using the CDS bed file, leaving you with this structure: 5’/ a hole corresponding to the cds/3’. To finish, use bedtools closest with this last file and the CDS bed file to get the closest features to the right of each cds. You get the 3’ of each cds this way. Use bedtools intersect on the original Isoscm file and you are left with only the 3’ features (exons and introns).”
In other words:
Exons = gene - introns
CDS = gene - introns - UTRs
therefore also:
CDS = Exons - UTRs
This makes sense to me. First, I’m going make a cds and exon specific gff file.
cd /data/putnamlab/jillashey/Astrangia_Genome
awk '$3 == "exon"' apoculata_v2.0.gff3 > exons.gff
wc -l exons.gff
239708 exons.gff
head exon.gff
chromosome_1 tRNAScan-SE exon 12926481 12926553 62.76 + . ID=Ser_58_exon;Parent=Ser_58_tRNA
chromosome_1 tRNAScan-SE exon 20472613 20472685 57.43 - . ID=Ser_104_exon;Parent=Ser_104_tRNA
chromosome_1 . exon 15863279 15863694 . - . ID=evm.model.chromosome_1.1448.exon3;Parent=evm.model.chromosome_1.1448
chromosome_1 . exon 15864523 15864816 . - . ID=evm.model.chromosome_1.1448.exon2;Parent=evm.model.chromosome_1.1448
chromosome_1 . exon 15865287 15865521 . - . ID=evm.model.chromosome_1.1448.exon1;Parent=evm.model.chromosome_1.1448
chromosome_1 . exon 15865882 15865901 . - . ID=evm.model.chromosome_1.1449.exon27;Parent=evm.model.chromosome_1.1449
chromosome_1 . exon 15866163 15866244 . - . ID=evm.model.chromosome_1.1449.exon26;Parent=evm.model.chromosome_1.1449
chromosome_1 . exon 15866990 15867097 . - . ID=evm.model.chromosome_1.1449.exon25;Parent=evm.model.chromosome_1.1449
chromosome_1 . exon 15867543 15867625 . - . ID=evm.model.chromosome_1.1449.exon24;Parent=evm.model.chromosome_1.1449
chromosome_1 . exon 15868562 15868656 . - . ID=evm.model.chromosome_1.1449.exon23;Parent=evm.model.chromosome_1.1449
awk '$3 == "CDS"' apoculata_v2.0.gff3 > cds.gff
wc -l cds.gff
236997 cds.gff
head cds.gff
chromosome_1 . CDS 15863279 15863694 . - 2 ID=cds.evm.model.chromosome_1.1448;Parent=evm.model.chromosome_1.1448
chromosome_1 . CDS 15864523 15864816 . - 2 ID=cds.evm.model.chromosome_1.1448;Parent=evm.model.chromosome_1.1448
chromosome_1 . CDS 15865287 15865377 . - 0 ID=cds.evm.model.chromosome_1.1448;Parent=evm.model.chromosome_1.1448
chromosome_1 . CDS 15865882 15865901 . - 2 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
chromosome_1 . CDS 15866163 15866244 . - 0 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
chromosome_1 . CDS 15866990 15867097 . - 0 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
chromosome_1 . CDS 15867543 15867625 . - 2 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
chromosome_1 . CDS 15868562 15868656 . - 1 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
chromosome_1 . CDS 15870067 15870167 . - 0 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
chromosome_1 . CDS 15870684 15870725 . - 0 ID=cds.evm.model.chromosome_1.1449;Parent=evm.model.chromosome_1.1449
There are multiple exons and CDSs for many genes. I’m going to merge both of these features with bedtools merge.
cd /data/putnamlab/jillashey/Astrangia_Genome
mkdir scripts
cd scripts
Bedtools merge requires that the data is pre-sorted by chromosome and then by start position. In the scripts folder: nano bed_merge.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Sorting by chromosome and then by start position" $(date)
sort -k1,1 -k4,4n exons.gff > exons.sorted.gff
sort -k1,1 -k4,4n cds.gff > cds.sorted.gff
echo "Sorting complete, starting merge with exons" $(date)
bedtools merge -i exons.sorted.gff > exon.merge.bed
echo "Exon merge complete, starting merge with CDS" $(date)
bedtools merge -i cds.sorted.gff > cds.merge.bed
echo "Merges complete!" $(date)
Submitted batch job 309744. That ran super fast but the number of lines in each file did not decrease all that much. I’m going to add the -d flag, which controls how close two features must be in order to merge. By default, it is set to 0, but I’m going to set it at 2000. Submitted batch job 309748. Once again, ran super fast.
wc -l exon.merge.bed
47035 exon.merge.bed
wc -l cds.merge.bed
46485 cds.merge.bed
These values make more sense to me, as that is about how many genes are in the genome. But they are still not identical…Going to do the subtraction anyway and see what happens. I’ll subtract the exon from the CDS? Using bedtools subtract. In the scripts folder: nano bed_subtract.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Subtracting exons from CDS" $(date)
bedtools subtract -a cds.merge.bed -b exon.merge.bed > test.bed
echo "Subtraction complete!" $(date)
Submitted batch job 309749. Test.bed file is empty…I’m guessing this was because the files are different lengths. I need to somehow remove the genes that do not have CDSs or exons annotated. In R (code here), I created a separate column for the parent ID in the attribute column and removed rows that do not have a row corresponding to an mRNA, an exon or a CDS. I then made two dataframes, one that contained only exons and one that contained only CDSs. I saved these as new gffs and put it in the Astrangia genome folder on Andromeda. I edited the bed_merge.sh to include the new gffs as input. Submitted batch job 309756. Look at number of lines and header:
wc -l exon.merge.bed
67105 exon.merge.bed
wc -l cds.merge.bed
64688 cds.merge.bed
head exon.merge.bed
Ala_1098_tRNA 22229936 22230008
Ala_1107_tRNA 20496372 20496446
Ala_1112_tRNA 18122334 18122406
Ala_1113_tRNA 18121779 18121851
Ala_1114_tRNA 18121543 18121615
Ala_1115_tRNA 18121306 18121378
Ala_1116_tRNA 18121069 18121141
Ala_1117_tRNA 18120832 18120904
Ala_1144_tRNA 11456062 11456134
Ala_1158_tRNA 8187155 8187227
head cds.merge.bed
evm.model.chromosome_10.1 12622 13741
evm.model.chromosome_10.10 55895 56373
evm.model.chromosome_10.10 58725 58982
evm.model.chromosome_10.100 966627 971367
evm.model.chromosome_10.1000 9548910 9549171
evm.model.chromosome_10.1001 9559190 9569583
evm.model.chromosome_10.1001 9572518 9574130
evm.model.chromosome_10.1002 9580187 9582242
evm.model.chromosome_10.1003 9587494 9595116
evm.model.chromosome_10.1004 9597669 9600588
They are still not the same number of lines. But let’s QC real quick. Pull out the bed data from a gene that is in both the exon and cds files.
# cds file
evm.model.chromosome_10.1004 9597669 9600588
# 9600588 - 9597669 = 2919
evm.model.chromosome_10.1004 9602637 9607464
# 9607464 - 9602637 = 4827
# exon file
evm.model.chromosome_10.1004 9597669 9600588
# 9600588 - 9597669 = 2919
evm.model.chromosome_10.1004 9602637 9607464
# 9607464 - 9602637 = 4827
Both the cds and exon file has two lines for this gene, interesting. Why? Maybe too far apart to be grouped even though they appear to be assigned to the same gene. If I subtract the end of the first entry from the start of the second entry, I get: 9607464 - 9597669 = 9795. So these two portions of the same gene (I think) are separated by ~10000 bp. I’m going to increase the -d flag to 15000. I also added wc -l into the script for each ending file so I don’t have to do it manually. Submitted batch job 309759. Still not the same, but I am going to remove any gene/chromosome names that have tRNA in them, as I don’t think these are true exons. Removed them in R and reuploaded gffs to server. Now rerunning bed_merge.sh. Submitted batch job 309764. Here are the line numbers.
46551 exon.merge.bed
46547 cds.merge.bed
So close… I wonder what the discrepancy is? The rows also look similar in both files…Changing d flag to 10000 and rerunning. Submitted batch job 309766
47001 exon.merge.bed
46993 cds.merge.bed
Still close…and still look the same:
head -20 exon.merge.bed
evm.model.chromosome_10.1 12622 13741
evm.model.chromosome_10.10 55895 58982
evm.model.chromosome_10.100 966626 971376
evm.model.chromosome_10.1000 9548910 9549171
evm.model.chromosome_10.1001 9559190 9574130
evm.model.chromosome_10.1002 9580187 9582242
evm.model.chromosome_10.1003 9587494 9595116
evm.model.chromosome_10.1004 9597669 9607464
evm.model.chromosome_10.1005 9616973 9637640
evm.model.chromosome_10.1006 9645049 9652090
evm.model.chromosome_10.1007 9660976 9662278
evm.model.chromosome_10.1008 9662508 9662954
evm.model.chromosome_10.1009 9663218 9665033
evm.model.chromosome_10.101 971989 975710
evm.model.chromosome_10.1010 9667643 9669122
evm.model.chromosome_10.1011 9669880 9673441
evm.model.chromosome_10.1012 9676142 9683173
evm.model.chromosome_10.1013 9689782 9691495
evm.model.chromosome_10.1014 9697732 9706569
evm.model.chromosome_10.1015 9707472 9711010
(base) [jillashey@ssh3 Astrangia_Genome]$ head -20 cds.merge.bed
evm.model.chromosome_10.1 12622 13741
evm.model.chromosome_10.10 55895 58982
evm.model.chromosome_10.100 966627 971367
evm.model.chromosome_10.1000 9548910 9549171
evm.model.chromosome_10.1001 9559190 9574130
evm.model.chromosome_10.1002 9580187 9582242
evm.model.chromosome_10.1003 9587494 9595116
evm.model.chromosome_10.1004 9597669 9607464
evm.model.chromosome_10.1005 9616973 9637640
evm.model.chromosome_10.1006 9645049 9652090
evm.model.chromosome_10.1007 9660976 9662278
evm.model.chromosome_10.1008 9662508 9662954
evm.model.chromosome_10.1009 9663218 9665033
evm.model.chromosome_10.101 971989 975710
evm.model.chromosome_10.1010 9667643 9669122
evm.model.chromosome_10.1011 9669880 9673441
evm.model.chromosome_10.1012 9676142 9683173
evm.model.chromosome_10.1013 9689782 9691495
evm.model.chromosome_10.1014 9697732 9706569
evm.model.chromosome_10.1015 9707472 9711010
If I subtract the cds from the exon, it will result in 0. Not sure what to do here…I may call it quits for the day here. Will ask the molecular mechanisms team tomorrow if they have any suggestions.
20240325
Javi sent me code to create 3’UTRs! I will use it to create 3’UTRs for my GFF so that I can extract the 3’UTR sequences for miranda. First, identify the counts of each feature from the gff file.
GFF_FILE="/data/putnamlab/jillashey/Astrangia_Genome/apoculata_v2.0.gff3"
genome="/data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta"
grep -v '^#' ${GFF_FILE} | cut -s -f 3 | sort | uniq -c | sort -rn > all_features.txt
cat all_features.txt
239708 exon
236997 CDS
47156 gene
45867 mRNA
44823 stop_codon
44312 start_codon
2889 five_prime_UTR
2317 tRNA
687 three_prime_UTR
Extract feature types and generate individual gffs for each figure. In Javi’s code, he has grep $'\tmRNA\t' ${GFF_FILE} | grep -v '^NC_' > ACER_k2.GFFannotation.mRNA.gff, which I will follow. I’m not sure why he is removing lines that have NC_ in them…maybe its a Acerv specific thing? I’m going to remove it for now, but may come back if I need to filter out specific rows.
grep $'\texon\t' ${GFF_FILE} > apoc_GFFannotation.exon.gff
grep $'\tCDS\t' ${GFF_FILE} > apoc_GFFannotation.CDS.gff
grep $'\tgene\t' ${GFF_FILE} > apoc_GFFannotation.gene.gff
grep $'\tmRNA\t' ${GFF_FILE} > apoc_GFFannotation.mRNA.gff
grep $'\tstop_codon\t' ${GFF_FILE} > apoc_GFFannotation.stop_codon.gff
grep $'\tstart_codon\t' ${GFF_FILE} > apoc_GFFannotation.start_codon.gff
grep $'\tfive_prime_UTR\t' ${GFF_FILE} > apoc_GFFannotation.five_prime_UTR.gff
grep $'\ttRNA\t' ${GFF_FILE} > apoc_GFFannotation.tRNA.gff
grep $'\tthree_prime_UTR\t' ${GFF_FILE} > apoc_GFFannotation.three_prime_UTR.gff
Extract chromosome lengths
cat is ${genome} | awk '$0 ~ ">" {if (NR > 1) {print c;} c=0;printf substr($0,2,100) "\t"; } $0 !~ ">" {c+=length($0);} END { print c; }' > apoc.Chromosome_lenghts.txt
cat apoc.Chromosome_lenghts.txt
chromosome_1 21106950
chromosome_2 21532011
chromosome_3 22725890
chromosome_4 27282911
chromosome_5 29602567
chromosome_6 30212294
chromosome_7 30683508
chromosome_8 29863146
chromosome_9 31044737
chromosome_10 33861578
chromosome_11 33534404
chromosome_12 42634786
chromosome_13 42928715
chromosome_14 58409227
Extract scaffold names
awk -F" " '{print $1}' apoc.Chromosome_lenghts.txt > apoc.Chromosome_names.txt
Sort the gffs by chromosome name. In the scripts of the Astrangia_Genome folder: nano bed_sort.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=5
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Sorting gffs by chromosome" $(date)
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.exon.gff > apoc_GFFannotation.exon_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.CDS.gff > apoc_GFFannotation.CDS_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.gene.gff > apoc_GFFannotation.gene_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.mRNA.gff > apoc_GFFannotation.mRNA_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.stop_codon.gff > apoc_GFFannotation.stop_codon_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.start_codon.gff > apoc_GFFannotation.start_codon_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.five_prime_UTR.gff > apoc_GFFannotation.five_prime_UTR_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.tRNA.gff > apoc_GFFannotation.tRNA_sorted.gff
sortBed -faidx apoc.Chromosome_names.txt -i apoc_GFFannotation.three_prime_UTR.gff > apoc_GFFannotation.three_prime_UTR_sorted.gff
echo "Sorting complete!" $(date)
Submitted batch job 309987. Ran super fast. Use flankBed to extract the UTRs around the genes. Javi used 2kb and 3kb as his cutoffs (ie he extracted the flanks that were 2000 and 3000 bp around his gene of interest). In his actual analysis, he used 3kb. I’m going to use 3kb for now, but may have to redo with a different number. I need to look into how far the UTRs are away from the gene end. I’m also going to remove portions of the UTRs that overlap with neighboring genes. In the scripts folder: nano flank_sub_bed.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=5
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Extracting 3kb UTRs" $(date)
flankBed -i apoc_GFFannotation.gene_sorted.gff -g ${genome} -l 0 -r 3000 -s | awk '{gsub("gene","3prime_UTR",$3); print $0 }' | awk '{if($5-$4 > 3)print $1"\t"$2"\t"$3"\t"$4"\t"$5"\t"$6"\t"$7"\t"$8"\t"$9}' | tr ' ' '\t' > apoc.GFFannotation.3UTR_3kb.gff
flankBed -i apoc_GFFannotation.gene_sorted.gff -g ${genome} -l 3000 -r 0 -s | awk '{gsub("gene","5prime_UTR",$3); print $0 }'| awk '{if($5-$4 > 3)print $1"\t"$2"\t"$3"\t"$4"\t"$5"\t"$6"\t"$7"\t"$8"\t"$9}'| tr ' ' '\t' > apoc.GFFannotation.5UTR_3kb.gff
echo "Subtract portions of UTRs that overlap nearby genes" $(date)
subtractBed -a apoc.GFFannotation.3UTR_3kb.gff -b apoc_GFFannotation.gene_sorted.gff > apoc.GFFannotation.3UTR_3kb_corrected.gff
subtractBed -a apoc.GFFannotation.5UTR_3kb.gff -b apoc_GFFannotation.gene_sorted.gff > apoc.GFFannotation.5UTR_3kb_corrected.gff
echo "UTRs identified!" $(date)
Submitted batch job 309994. Gave me this error:
*****ERROR: Unrecognized parameter: 0 *****
*****ERROR: Need both -l and -r.
*****ERROR: Must supply -l and -r or just -b with -s.
It does not seem to like 0 being set for -l or -r. I’m going to set the 0 to 1 instead. Submitted batch job 309995. Still getting an error saying unrecognized parameter. Replace flankBed with bedtools flank and subtractBed with bedtools subtract. Submitted batch job 309997. Still getting an error saying unrecognized parameter. I might need to give the -g flag the apoc.Chromosome_lenghts.txt file. In the help notes, it says:
The genome file should tab delimited and structured as follows:
<chromName><TAB><chromSize>
For example, Human (hg19):
chr1 249250621
chr2 243199373
...
chr18_gl000207_random 4262
Going to add apoc.Chromosome_lenghts.txt as the -g argument. Submitted batch job 310001. Now getting the error: Error: Unable to open file apoc.GFFannotation.5UTR_3kb.gff. Exiting. Editing output file names so that they correspond to 5’ or 3’. Submitted batch job 310005. Worked, hooray! Now I will download the following files to my local computer: apoc.GFFannotation.3UTR_3kb_corrected.gff, apoc.GFFannotation.5UTR_3kb_corrected.gff, apoc_GFFannotation.gene_sorted.gff, and apoc_GFFannotation.mRNA_sorted.gff. After working through the R script that Javi provided me, I don’t think I need to do all of the R code that he proposed. I think I just need to extract the 3’UTR sequences from the genome using bedtools (example here). I can use bedtools getfasta. In the /data/putnamlab/jillashey/Astrangia_Genome/scripts folder: nano bed_getfasta_3UTR.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=5
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Extracting 3' UTR sequences" $(date)
bedtools getfasta -fi apoculata.assembly.scaffolds_chromosome_level.fasta -bed apoc.GFFannotation.3UTR_3kb_corrected.gff -fo apoc_3UTR.fasta -name
echo "Sequence extraction complete!" $(date)
Submitted batch job 310035. Ran in 9 seconds. Look at the output fasta.
zgrep -c ">" apoc_3UTR.fasta
60613
head apoc_3UTR.fasta
>3prime_UTR::chromosome_1:17663-20663
GGAACGCGTACCAATTTTATCCGTGATCGGACTACCTCGCGACTAGGTCCGGGTAAAATTTTGGTTCAGCACGGGTGAGGTAAATCCGTTTACACGTCAAATGTTATCCGTGCTGAACCATTTTTTTCGGTATGCGTTCCATTTTTACTCGAGCCGTGCCGCGTAAAATGGGCTTGAGTTATAAGCAATCAAAACTAAAATGAAGGGTGTCATTGCAAGATTAAACTGTTGCTTTGGTAACCTTTTATGTCATAAAAATCATAACAACGTGTTCCGCAATCATTGGGCATTTGTTTGACACCATGATTGTAGCATCAACTGATGAAGAGTGGTTAAAGTGACCCGTCAAAATCCAAGACTTGGAAAGTGCTGGAAAATTGAGCCAGCCACCTTAAACCTTCATCTTAACTCAGTGAAGGCAAGAACTAGCCAAAAATTCTTATAAACAATGAACAGGGTCAGTGACAGACCCCGTTAAGTGTCAACAGGATAACCCCCGACTACTCGTATTTATTATTCAAACAGTTCAGTTTCTCTCTAAGATAAAATTCAAATTTAAGAGGCTGTGTCTGTAATACTGGTCTGCCCGATCTGGCTTGTTAAATAGATTTCGCTCTTTTCTAGTGTGTTGCAAGACTTTCATGTACTTATACTAAAAACACAATTCACTTGACCGGATCATTAATTTTTTTTTCGAGTTTTTAAGGATTTTCGATTCACCCGAACGAGCACTTGTAACGACCACTGTCAAGGCTTAAATTTACCACAACTTTGAAAATTAATTAAAACAGAACTAACAACTTCTGTACCAAACAAACACCACAGCCTGTTAAATTATGTCTGTTTAATTTATCACACCAACTTTCACGAACATTGAGCTCGAATCAAGCTAGCAGCAAAACATTAACGACACCCCTATCACACATCTTGATGAAACACTTGAAAACGGCTCAGATTCAAACTTTAGTTTTGAAAAAGTGGGGCGGTCTACTTCAATCAAACCAACGCAGCCGAAAAGTAGATGTAAAAAAAAAATGCTCCTATCGTAAAAAACCTTCTGGTATCGTCTTAATGACATATTTACAGCTGTGTAAACTTGCCCAATTTCGAAGATCTTCATGTTTGCAGTCATTTTGAGGGACTCGTCAGGACGAGGAGTTTGATCAACTGGCCTCGCAGACATACCTAACACCATCAGAAATAAACAAAGATTAGATATAAATGCATTGCAGTTCAACTGAAAGAGAATAAATCGTGTAATAGATCAACGGGTCGAGAATTATGATTAACGTTTAGCTATAAGCTTTATATTGATCTACTCCCGTTGTTATTGTTGAATGAATCGGAGAGTTTTTCAAAAAATTATTTAACATCTGTTACAAATAAAAATAACAACAATTTTTTTTTTCAAGTTTACAAAATAGCTCGTGCAGTATTTGATCTTAACGACTGAAAAAGACATAGTATATCTTGTTTCTAAGCCCTTACCTCTTTTATCGGGTCGAGAATTCACCTTATAAAACTCTGTGCGTTTGCAAATAAAACAAAACAAATCATTTCGCACTGATCAAGCTTGACGCGTTGTTTAAATGCCGCCGACATCGAAAAAATTACACCAATATTGTAGCCATCAGATACCATCTGTGGCGTCCGAAATATATTATCGTAAGTAAAGAAATAGTTTATTCTATGTTCGTTTTTAAAAAAGATTAATAAAAATGAAATCGTTTTCAGTAAACCTACCAAAGTTTCGTGATGTTAACGTTCCCTGATGGTGTTCAATTATTCTAAAAATAGTCGTTTAATTTGAACGGAATAGTTTCAATTAAAACTTAAATATCTTTTAATTTTAAAAAAGTTAATATACAAGGCTTCCAAAAATAAACAAACCAGCATGCACAGAACATTTTTTAGCAAAACTGAACGAGTTTTTCTGGGTAGGGTTATAAAACAGGCTACGCCTGCACTGTTATAATGAAACGCTTTGTTAAAGGGAACCTCTGTTGTTTAGGCAAGTTGACGTTGAGTTGTTTGAAATAATGCCTAAACTACAATTGTACAGGACAGGGGAGAAACAGCATTTAATAACAACCGTAAACGGAAAAGAAAAAGGCTTTTAAATTTCCCGCTCGGCGCCATTTTGAAATTTATGACGTTAGTGCGTTGTCCAAATGTTTCACTCAAAAGAACCTCTGCTTTCTAATAATAGCGGCAACGCAGTGACGTCATAAAATTCAAAATGGCGGCGCGCGGGAAATTCAAAAGCACAAAAATTGACAAATCAAAGGCCCAGTATTCCAATTAAAAAATGGAATCCGCGACTTTAAACAACCAAAACGATTTTAGACTGTGGTAAACTTATTATTTTGACCAAAACTGTTTTACAGTCGAGGTTCCCTTTAAGCAAAACACCTTAAAGCTTAATAGTCCAGTCATTTTGTGGTTGGACCTGCAACTAATTGCAACAGAAGCTTTTTGACCGCAAATCATTTCGAATAAGTCTCTAGATTAACATACTAAAAGTCCCAAAGGATTCATGCATGGGAACATCTCGAAAATTTAACGGATTCTAAGTTGTGCTTTTGCGTAACATGAAAACGAGGCTCAAAAACAGAGGTTCTGTTATTATGCTTGTTTATGGCAAGAATTTCATTTATTTCATCCAAAAACAATGAAATATACTTTAAATATGCTTGAAAACTTTCATCCAGAGGAAAAATTAATCAAAAAGTATTCAATAAGCAAAATAAATCTCATTATATTTAAATTTTCAATTGTGAAGCGAATTGGACTACACCAAAAATGGTAGTTGCGATAGTTTATTATCCTTTTATGGATCGCACAAAAAAATTGCAAGCGAGCCAACAAATAAATATATCTTTCACAATGGTGAAATATTCTAAAAGAACATCGTTTGTGAAATGAAAACGAATTTCACAGTTTCTTCCAGCTCTTGTGTCAATCACCGTCGTCATCGCTATCATCGAAATCTTGATGGGG
>3prime_UTR::chromosome_1:40489-43489
TTCCAGGAATTTTGAAATAATTTTCTACAATTAATTTATTGTAATAAGAAAATCAACTGTATGCAATTGTTATGTAGACAGTCTCTAAATATCTTCTTAGTACACTCCAGAAAGCACATCATTTCTCACCACCATTGAAAAGTGCATATAAAAACTCTCAGATTCTCGCCATGTTTTCGAGCTGCAAAGAGCTTTATAACTTCTTTTGGATCCTCTCCGCATATTCGTGACGTAAATCTGATTTCACAGGTAGCCCAGGAGGTCTGAACTCCGCTTTCACTGTAGTCGTGTTCGGGTGGATTTTGTCTTGATTCAAAATACTAGCTGGTGTGGCTTGTCCAGCTGGATGTAAAGTTTATATATAGCTTTTTAGAACCAGAAACACAACTAGTATCTATTATTTTATACACTTTTTAATTGTGGCGGGAAAACGTGTACAACATGATGTACGTTGATGGGATCGTGTCCTCAAAGACCCACTGCCCCAAGGCCATGGAGTCCAAAGCACGGCCCCTATATATTTGTTTTAATCTTCAAGGCAAATGCGCATTACCAGTATCAATTATCAAGGATGGACCGTGAACAGTGTAGAAATAAACAGAGTAAGAAACGAAACAAAGTATTTCCTTGTTCCTATGCTTTATGGACATTGGTGGCTTGATTATACTTTATCTTCTTAATCTGGACAGTTGAGGTTTCGCAAGAAGTGTTTAAGTGAATGTTTACCTGTTCATGAGGGTGATCAGACCGCTTTTTGTAGCAGAATTTAATGCCAGATATTTTCATTGATTTCCGTCCGCCATGTTGGAGCCCAAGCAGTTGGGCTCCAACATGGCGTCTCCATAATGTGCTCTCTAAATTTGCGTGAAACATTTTGACGGATAACTCGAGTACGAAATAATGTAAAGACCTGAGACTTGGACAAGTAGTATGTTTATATAGTAGTTTTCTACAACCTGTAATATTCTTGATTTTTTTGACTCAATAGTTTTGGATTTTTTTTCGCGGCGTGACAGTGAAAACCACCTATTGAAATGGATCGCGCCTTCGGCACCCGAGGAGTTTAATTCACGTTGCCCCCGAATAGAGAGCCTGCTGGCAGGCTACAGTTAATCTTAAGTTGCATTATGTAACCGGGCCCTGGCCCAGTTCTTCGTGTTCCACTTGTATTTGAGTGATTTGTTGGTCTTTGTTTGCCAAAAACAAAAGAAAAAAATCAAAAATGTCGAGCAAAATCTAAATTATAGACTACGTATAGAACACAATAGTGGCTAAAGCGTAAAAATGGCCAGTTACACAGCGACCTCACACAAGAGTGCATGGACGGCGGAGCATTTTTCCAACTGGGGGGGCTTATGAGACTCTTCTCTTATAAATTCTTTATTTTTTCTGTAAAAGTGGGGGGCTAAAGCCCCCCCAGCCCCCCTGGCTCCGCCGTCCCTGGAGTGTTTGGGAAATAGAGATTACTCCATGGAAAATGCGCGCGTACGATTTTTATTCACGAGTTGAGTAGTATCAGAAAACGAACGAGTGAGCGTAGCGAACGAGTGATTTTTCTGATACAACTCTGCGAGTGAATAAAAATCGTACAAAGCATTTTCCATGGTGTAATTTGTTTATTTTATAGATACTGAGGTTTTTGGAAAGCAAATTAAATCGCTCGACTTTTTATCAGTTACGCGCGTGTTTAGGAAACCCATTCATCTGCCATAAACTAAAATAACTAAAACATGTTTGTCTTTCAAAATCACATCTTTAATGAAGTGTAGCACAAGAAATAAGACGTCAGTTTATACTCTCGCAACTTTTACAGAAACAACTTTTGATAGAAAACCTGACTGAGACTTGTACAAAACTTGCAAGTTCCAGTTCAATGTTTTAACAGAACAAAACAGCGCTAAACGCTGAATAACTCATCGCGAACTCGAGCGAACTTTTACAAATTAAATCAAGAAAACAGTCTGCCAAAACCAAGGACAAACCTGCAGTGTTTTAGTCATCATCTGAATCGAAACTCGCTGCAATCCCTCGGAGTGAAGACGGCCCGTATTCTTGTCCTTCCTTTGTGCGAATTTATACTCAAAGGTTTATTCAACTCGGCAGGAGGAATTTCGGTTATCTCGCTTCGTGTTTCTCTTTTTGTTTGAAGAAACGTTGTGACAACAAGGCCGTCTTGTTTCGTTTTCTTTATAGTGTTTTGGTTTTCCTGTTCTGCAATATAAACGACTCAGTGGAGCCGTTAAAGACACAAACAAACCTTGAACGTGTCTCCGCCATGTCGCCGGTGTTTACGACGAGAATCAAAATGGCGATACGGTGTTGTTCACAAGTGAGTTTTGGCCGTTCGCGCTTGTGATTGGACGAACGACGTTTTTTCACTAGTGAGTTTTCATCGTTCGCCTTTCCTATTGGCTAATGCATTTTACGTATCATTTGTACGCAATCGTTTTTTCGTGAGAAAATCTCAGTATCTATGATATAAATAGTTGTTCTGTTCTGATATTTCCATCGGTTTGTATAAAAATAACCTAAGAAGGCGAGAAACAAAACTGTTACGAATCATTTACTTTTCTAGCCCCAAAAGATGATTCCCAGGGGTTTTTGCCTCGTTCTTGACTTAGAGGGGTGTGGTACTTGACCGATGTTTGGTTATTAGGGTGTTACAGAGGGTTTTTAAATGTTCAAAGACTTCCTAAGGAAGCTCCCTCTCTTTACGGACACGGACACCTCCCTGTTAAAGACAGTTTATTTGGTCCGTAAGAGATCAGAATTTATACAAACTTTACCTCTGTAATACGAATGCCTCCATGATCTAGCCGCTTTTTTCTAACCCTTTGGTATCCGACCGCGCTATTTTTTGTAACGTTTCAAACGCGCGAAGGACAATCGAAGCTTCTCGTACGATGAACGTATAATTTCTCAAAAAACTAGCTTCATGTACTTCATTTTCATTCCCAGGCCTTCAAACTGACAGAATATGAGCAAAATCGTTTTATGAAT
>3prime_UTR::chromosome_1:53463-55801
Hooray! I have the 3’ UTRs! But how do I know which gene they go with?? I guess the location? Editing the script above so that instead of -name, its fullHeader. Submitted batch job 310037. Nope that didn’t change anything. Well somehow I need to figure out how to obtain the gene id that the 3’UTR sequence corresponds to, likely based on the coordinates of the 3’UTR and gene sequences.
20240326
I’m going to run a test of miranda with the mature miRNAs and the 3’UTRs. In the miranda documentation, file1 is the miRNA file and file2 is the 3’UTR sequences. There are options to play with scaling and alignment parameters (ie -en, -sc, -scale, -loose, etc) but for now I will just run a basic test to see if it works. In the scripts folder, nano miranda_test.sh:
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa /data/putnamlab/jillashey/Astrangia_Genome/apoc_3UTR.fasta
conda deactivate
miranda conda stuff appears to be empty on the server…might need to reactivate? Removing miranda from /data/putnamlab and going to try to create a new one in the conda folder on putnam lab.
cd /data/putnamlab/conda
module load Miniconda3/4.9.2
# Create a new environment
conda create --prefix /data/putnamlab/conda/miranda
conda install miranda
Gives me this error: EnvironmentLocationNotFound: Not a conda environment: /data/putnamlab/miranda. There is a miranda folder but it only has the history of the commands that I have run for this. I deleted this folder and am going to try installing with mamba, as this is what the bioconda website recommends.
module load Mamba/22.11.1-4
# Create a new environment
mamba create --name miranda_JA miranda
Didn’t work, gave me this error:
Looking for: ['miranda']
error libmamba Could not open lockfile '/opt/software/Mamba/22.11.1-4/pkgs/cache/cache.lock'
error libmamba Could not open lockfile '/opt/software/Mamba/22.11.1-4/pkgs/cache/cache.lock'
conda-forge/noarch 16.3MB @ 7.2MB/s 2.4s
conda-forge/linux-64 39.1MB @ 7.1MB/s 6.2s
Could not solve for environment specs
Encountered problems while solving:
- nothing provides requested miranda
The environment can't be solved, aborting the operation
Trying again
module purge
module load Miniconda3/4.9.2
conda create --prefix /data/putnamlab/conda/miranda
cd /data/putnamlab/conda/miranda
conda install bioconda::miranda
Downloading and Extracting Packages
ca-certificates-2024 | 127 KB | ######################################################################################## | 100%
certifi-2024.2.2 | 159 KB | ######################################################################################## | 100%
miranda-3.3a | 58 KB | ######################################################################################## | 100%
Preparing transaction: done
Verifying transaction: failed
EnvironmentNotWritableError: The current user does not have write permissions to the target environment.
environment location: /opt/software/Miniconda3/4.9.2
uid: 1050
gid: 1012
Says that I do not have permissions to write to Miniconda location. But I’m not trying to write there. Asked chatGPT and this is what it gave me:
conda create --prefix /data/putnamlab/conda/miranda
conda activate /data/putnamlab/conda/miranda
conda install bioconda::miranda
Success! Looks like I just had to activate the environment. Let’s now trying running miranda. In the scripts folder, nano miranda_test.sh:
#!/bin/bash -i
#SBATCH -t 72:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa /data/putnamlab/jillashey/Astrangia_Genome/apoc_3UTR.fasta
conda deactivate
Submitted batch job 310076
20240328
miranda test finished in about a day. The output is in slurm-310076.out in the scripts folder, I should’ve piped it to an output file but oh well. The file is quite large. Here’s an example of the information in it:
Read Sequence:chromosome_1:40489-43489 (3000 nt)
=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=
Performing Scan: chromosome_12_481048 vs chromosome_1:40489-43489
=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=
Forward: Score: 152.000000 Q:2 to 18 R:855 to 875 Align Len (17) (70.59%) (82.35%)
Query: 3' cccTTGTTCG-GCTTTGTAAc 5'
||: || :||||||||
Ref: 5' ctaAATTTGCGTGAAACATTt 3'
Energy: -11.470000 kCal/Mol
Scores for this hit:
>chromosome_12_481048 chromosome_1:40489-43489 152.00 -11.47 2 18 855 875 17 70.59% 82.35%
Score for this Scan:
Seq1,Seq2,Tot Score,Tot Energy,Max Score,Max Energy,Strand,Len1,Len2,Positions
>>chromosome_12_481048 chromosome_1:40489-43489 152.00 -11.47 152.00 -11.47 2 20 3000 855
Complete
So much information…Let’s go through this line by line. The top line Read Sequence is the 3’UTR sequence. The Performing Scan line indicates the specific miRNA that will be compared to the specified 3’UTR sequence. The Forward line has information about the score and the lengths of the query and reference sequences that matched up. I’m not sure what the percentages mean…Maybe its a percent of base pairs in the miRNA that aligned? The Query is the miRNA sequence and the Ref is the 3’UTR sequence. The bars between the query and reference sequence represent aligned bps and the dots between the sequences an instance of a base wobble pair. The Scores for this hit has the total score and energy of this interaction. A good cutoff is typically 100-150 for score and -10 kcal/mol for energy. I think the 2 and 18 is the miRNA sequence that aligns with the query, and vice versa with the 3’UTR sequence (855 and 875). Still not sure what the percentages mean. The Scores for this scan includes much of the same information as above. I’m guessing that hit vs scan is the hit of the miRNA to a specific portion of the 3’UTR and the scan refers to all of the hits along a specific 3’UTR sequence? Not totally sure though.
This file is massive. It would have taken ~1 hour to download to my personal computer. I’m going to add some lines to the script that count all of the putative interactions. This code is being added to the miranda_test.sh script (and the actual miranda code is being commented out so it does not run again).
zgrep -c "Performing Scan" slurm-310076.out
I used the “Performing Scan” phrase because I believe that this is the header for each putative interaction. Submitted batch job 310209. Ran in about 6 mins and there are 112,558,341 putative interactions! That is crazy. I think I should apply some filtering parameters in the miranda code now. I’m going to add -sc 100 and -en -10. Also adding -out test_miranda.tab. Submitted batch job 310211
20240401
Miranda has pre-emptively stopped and restarted a couple times. I’m going to stop job 310211 now and assess the results thus far.
These were the current settings:
Current Settings:
=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=
Query Filename: /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa
Reference Filename: /data/putnamlab/jillashey/Astrangia_Genome/apoc_3UTR.fasta
Gap Open Penalty: -9.000000
Gap Extend Penalty: -4.000000
Score Threshold: 100.000000
Energy Threshold: -10.000000 kcal/mol
Scaling Parameter: 4.000000
If I want to invoke strict seed binding, I will probably have to play with the gap open and extend penalty thresholds. Here’s an example of one of the miRNA:mRNA matches:
Performing Scan: chromosome_12_481048 vs chromosome_1:17663-20663
=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=
Forward: Score: 143.000000 Q:2 to 17 R:2880 to 2900 Align Len (16) (75.00%) (87.50%)
Query: 3' ccctTGTT-CGGCTTTGTAAc 5'
|||| | :||||:|||
Ref: 5' tttcACAATGGTGAAATATTc 3'
Energy: -12.000000 kCal/Mol
Scores for this hit:
>chromosome_12_481048 chromosome_1:17663-20663 143.00 -12.00 2 17 2880 2900 16 75.00% 87.50%
Very cool! How do I invoke strict seed binding?
20240409
In order for miranda to run faster, I am going to filter the input so that I’m only getting the differentially expressed genes and miRNAs. First, I’m going to subset the mature_all.fa file. This is a list of my differentially expressed miRNAs. I’m copying it to the server into /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57. Subset the mature fasta file based on the miRNAs in the DEM list.
grep -A 1 -f DEM_list.txt mature_all.fa | grep -v "^--$" > mature_DE.fa
zgrep -c ">" mature_DE.fa
50
Excellent, now all differentially expressed miRNAs are in the mature_DE.fa. Time to work on subsetting the gene list by the DEGs. This one is a little bit more complicated because I have a fasta of the 3’UTR sequences for each gene which is being used as input for the miranda script. However the 3’UTR sequences are only labeled with a coordinate (ie >chromosome14:1-200) as opposed to what gene corresponds to that specific 3’UTR sequence (ie >gene1). I can do this with bedtools intersect (also used above). First, convert the fasta headers of the 3’UTR fasta file to bed file format.
cd /data/putnamlab/jillashey/Astrangia_Genome
awk -F'[:-]' '/^>/ {print $2 "\t" $3-1 "\t" $4}' apoc_3UTR.fasta > apoc_3UTR.bed
Intersect BED files with primary GFF. In the scripts folder: nano bed_intersect_3UTR.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Merging 3'UTR bed file with gff file to get genes associated with 3'UTR sequences" $(date)
bedtools intersect -a apoc_3UTR.bed -b apoculata_v2.0.gff3 -wa -wb > intersect_output.txt
echo "Merge complete" $(date)
Submitted batch job 310666. Got this error:
Error: Type checker found wrong number of fields while tokenizing data line.
Perhaps you have extra TAB at the end of your line? Check with "cat -t"
Apparently the bed tools is looking for the bed file to have chromosome, start and end columns. I need to include the chromosome columns
awk -F'[:-]' '/^>/ {chromosome=substr($1, 2); start=$2; end=$3; print chromosome "\t" start "\t" end}' apoc_3UTR.fasta > apoc_3UTR.bed
head apoc_3UTR.bed
chromosome_1 17663 20663
chromosome_1 40489 43489
chromosome_1 53463 55801
chromosome_1 53463 55801
chromosome_1 74713 76735
chromosome_1 74713 76735
chromosome_1 92518 93919
chromosome_1 94852 95518
chromosome_1 92518 93919
chromosome_1 101190 104190
Now try running the bed_intersect_3UTR.sh script. Submitted batch job 310668. It runs but the output file is 0…try using apoc_GFFannotation.gene_sorted.gff as gff input. Submitted batch job 310669. Still produced nothing…
head apoc_GFFannotation.gene_sorted.gff
chromosome_1 EVM gene 20664 21393 . - . ID=evm.TU.chromosome_1.1;Name=EVM%20prediction%20chromosome_1.1
chromosome_1 . gene 34636 40489 . + . ID=evm.TU.chromosome_1.2;Name=EVM%20prediction%20chromosome_1.2
chromosome_1 . gene 43758 53463 . + . ID=evm.TU.chromosome_1.3;Name=EVM%20prediction%20chromosome_1.3
chromosome_1 . gene 55802 60431 . - . ID=evm.TU.chromosome_1.4;Name=EVM%20prediction%20chromosome_1.4
chromosome_1 . gene 62282 74713 . + . ID=evm.TU.chromosome_1.5;Name=EVM%20prediction%20chromosome_1.5
chromosome_1 . gene 76736 78519 . - . ID=evm.TU.chromosome_1.6;Name=EVM%20prediction%20chromosome_1.6
chromosome_1 . gene 78661 92518 . + . ID=evm.TU.chromosome_1.7;Name=EVM%20prediction%20chromosome_1.7
chromosome_1 . gene 93920 94852 . - . ID=evm.TU.chromosome_1.8;Name=EVM%20prediction%20chromosome_1.8
chromosome_1 . gene 104191 109764 . - . ID=evm.TU.chromosome_1.9;Name=EVM%20prediction%20chromosome_1.9
chromosome_1 . gene 113080 113418 . - . ID=evm.TU.chromosome_1.10;Name=EVM%20prediction%20chromosome_1.10
>chromosome_1:17663-20663
GGAACGCGTACCAATTTTATCCGTGATCGGACTACCTCGCGACTAGGTCCGGGTAAAATTTTGGTTCAGCACGGGTGAGGTAAATCCGTTTACACGTCAAATGTTATCCGTGCTGAACCATTTTTTTCGGTATGCGTTCCATTTTTACTCGAGCCGTGCCGCGTAAAATGGGCTTGAGTTATAAGCAATCAAAACTAAAATGAAGGGTGTCATTGCAAGATTAAACTGTTGCTTTGGTAACCTTTTATGTCATAAAAATCATAACAACGTGTTCCGCAATCATTGGGCATTTGTTTGACACCATGATTGTAGCATCAACTGATGAAGAGTGGTTAAAGTGACCCGTCAAAATCCAAGACTTGGAAAGTGCTGGAAAATTGAGCCAGCCACCTTAAACCTTCATCTTAACTCAGTGAAGGCAAGAACTAGCCAAAAATTCTTATAAACAATGAACAGGGTCAGTGACAGACCCCGTTAAGTGTCAACAGGATAACCCCCGACTACTCGTATTTATTATTCAAACAGTTCAGTTTCTCTCTAAGATAAAATTCAAATTTAAGAGGCTGTGTCTGTAATACTGGTCTGCCCGATCTGGCTTGTTAAATAGATTTCGCTCTTTTCTAGTGTGTTGCAAGACTTTCATGTACTTATACTAAAAACACAATTCACTTGACCGGATCATTAATTTTTTTTTCGAGTTTTTAAGGATTTTCGATTCACCCGAACGAGCACTTGTAACGACCACTGTCAAGGCTTAAATTTACCACAACTTTGAAAATTAATTAAAACAGAACTAACAACTTCTGTACCAAACAAACACCACAGCCTGTTAAATTATGTCTGTTTAATTTATCACACCAACTTTCACGAACATTGAGCTCGAATCAAGCTAGCAGCAAAACATTAACGACACCCCTATCACACATCTTGATGAAACACTTGAAAACGGCTCAGATTCAAACTTTAGTTTTGAAAAAGTGGGGCGGTCTACTTCAATCAAACCAACGCAGCCGAAAAGTAGATGTAAAAAAAAAATGCTCCTATCGTAAAAAACCTTCTGGTATCGTCTTAATGACATATTTACAGCTGTGTAAACTTGCCCAATTTCGAAGATCTTCATGTTTGCAGTCATTTTGAGGGACTCGTCAGGACGAGGAGTTTGATCAACTGGCCTCGCAGACATACCTAACACCATCAGAAATAAACAAAGATTAGATATAAATGCATTGCAGTTCAACTGAAAGAGAATAAATCGTGTAATAGATCAACGGGTCGAGAATTATGATTAACGTTTAGCTATAAGCTTTATATTGATCTACTCCCGTTGTTATTGTTGAATGAATCGGAGAGTTTTTCAAAAAATTATTTAACATCTGTTACAAATAAAAATAACAACAATTTTTTTTTTCAAGTTTACAAAATAGCTCGTGCAGTATTTGATCTTAACGACTGAAAAAGACATAGTATATCTTGTTTCTAAGCCCTTACCTCTTTTATCGGGTCGAGAATTCACCTTATAAAACTCTGTGCGTTTGCAAATAAAACAAAACAAATCATTTCGCACTGATCAAGCTTGACGCGTTGTTTAAATGCCGCCGACATCGAAAAAATTACACCAATATTGTAGCCATCAGATACCATCTGTGGCGTCCGAAATATATTATCGTAAGTAAAGAAATAGTTTATTCTATGTTCGTTTTTAAAAAAGATTAATAAAAATGAAATCGTTTTCAGTAAACCTACCAAAGTTTCGTGATGTTAACGTTCCCTGATGGTGTTCAATTATTCTAAAAATAGTCGTTTAATTTGAACGGAATAGTTTCAATTAAAACTTAAATATCTTTTAATTTTAAAAAAGTTAATATACAAGGCTTCCAAAAATAAACAAACCAGCATGCACAGAACATTTTTTAGCAAAACTGAACGAGTTTTTCTGGGTAGGGTTATAAAACAGGCTACGCCTGCACTGTTATAATGAAACGCTTTGTTAAAGGGAACCTCTGTTGTTTAGGCAAGTTGACGTTGAGTTGTTTGAAATAATGCCTAAACTACAATTGTACAGGACAGGGGAGAAACAGCATTTAATAACAACCGTAAACGGAAAAGAAAAAGGCTTTTAAATTTCCCGCTCGGCGCCATTTTGAAATTTATGACGTTAGTGCGTTGTCCAAATGTTTCACTCAAAAGAACCTCTGCTTTCTAATAATAGCGGCAACGCAGTGACGTCATAAAATTCAAAATGGCGGCGCGCGGGAAATTCAAAAGCACAAAAATTGACAAATCAAAGGCCCAGTATTCCAATTAAAAAATGGAATCCGCGACTTTAAACAACCAAAACGATTTTAGACTGTGGTAAACTTATTATTTTGACCAAAACTGTTTTACAGTCGAGGTTCCCTTTAAGCAAAACACCTTAAAGCTTAATAGTCCAGTCATTTTGTGGTTGGACCTGCAACTAATTGCAACAGAAGCTTTTTGACCGCAAATCATTTCGAATAAGTCTCTAGATTAACATACTAAAAGTCCCAAAGGATTCATGCATGGGAACATCTCGAAAATTTAACGGATTCTAAGTTGTGCTTTTGCGTAACATGAAAACGAGGCTCAAAAACAGAGGTTCTGTTATTATGCTTGTTTATGGCAAGAATTTCATTTATTTCATCCAAAAACAATGAAATATACTTTAAATATGCTTGAAAACTTTCATCCAGAGGAAAAATTAATCAAAAAGTATTCAATAAGCAAAATAAATCTCATTATATTTAAATTTTCAATTGTGAAGCGAATTGGACTACACCAAAAATGGTAGTTGCGATAGTTTATTATCCTTTTATGGATCGCACAAAAAAATTGCAAGCGAGCCAACAAATAAATATATCTTTCACAATGGTGAAATATTCTAAAAGAACATCGTTTGTGAAATGAAAACGAATTTCACAGTTTCTTCCAGCTCTTGTGTCAATCACCGTCGTCATCGCTATCATCGAAATCTTGATGGGG
>chromosome_1:40489-43489
TTCCAGGAATTTTGAAATAATTTTCTACAATTAATTTATTGTAATAAGAAAATCAACTGTATGCAATTGTTATGTAGACAGTCTCTAAATATCTTCTTAGTACACTCCAGAAAGCACATCATTTCTCACCACCATTGAAAAGTGCATATAAAAACTCTCAGATTCTCGCCATGTTTTCGAGCTGCAAAGAGCTTTATAACTTCTTTTGGATCCTCTCCGCATATTCGTGACGTAAATCTGATTTCACAGGTAGCCCAGGAGGTCTGAACTCCGCTTTCACTGTAGTCGTGTTCGGGTGGATTTTGTCTTGATTCAAAATACTAGCTGGTGTGGCTTGTCCAGCTGGATGTAAAGTTTATATATAGCTTTTTAGAACCAGAAACACAACTAGTATCTATTATTTTATACACTTTTTAATTGTGGCGGGAAAACGTGTACAACATGATGTACGTTGATGGGATCGTGTCCTCA
Hmm so the gene and the utr do not overlap by the looks of it. The first line in the sorted gene gff has start and stops here: 20664 21393 but the 3’UTR sequence goes from chromosome_1:17663-20663 so there is no overlap. Maybe I need to use the 3’UTR sorted gff file aka apoc_GFFannotation.three_prime_UTR_sorted.gff? Submitted batch job 310671. Still empty :’(. Maybe I calculated 3’UTR incorrectly…or maybe not but I am not getting the overlap because I used flank bed. so how to connect them…
This is what flank bed did:
I need to know the flanks, which are the 3’UTRs, and what is being flanked, which is the input or the gene itself. I need to know the genes that were flanked! Trying the apoc.GFFannotation.3UTR_3kb.gff. Submitted batch job 310686. This was the gff that was produced from flank bed. This got an output file but I’m not sure I understand it.
wc -l test.txt
117033 test.txt
head test.txt
chromosome_1 17663 20663 chromosome_1 EVM 3prime_UTR 17664 20663 . - . ID=evm.TU.chromosome_1.1;Name=EVM%20prediction%20chromosome_1.1
chromosome_1 40489 43489 chromosome_1 . 3prime_UTR 40490 43489 . + . ID=evm.TU.chromosome_1.2;Name=EVM%20prediction%20chromosome_1.2
chromosome_1 53463 55801 chromosome_1 . 3prime_UTR 53464 56463 . + . ID=evm.TU.chromosome_1.3;Name=EVM%20prediction%20chromosome_1.3
chromosome_1 53463 55801 chromosome_1 . 3prime_UTR 52802 55801 . - . ID=evm.TU.chromosome_1.4;Name=EVM%20prediction%20chromosome_1.4
chromosome_1 53463 55801 chromosome_1 . 3prime_UTR 53464 56463 . + . ID=evm.TU.chromosome_1.3;Name=EVM%20prediction%20chromosome_1.3
chromosome_1 53463 55801 chromosome_1 . 3prime_UTR 52802 55801 . - . ID=evm.TU.chromosome_1.4;Name=EVM%20prediction%20chromosome_1.4
chromosome_1 74713 76735 chromosome_1 . 3prime_UTR 74714 77713 . + . ID=evm.TU.chromosome_1.5;Name=EVM%20prediction%20chromosome_1.5
chromosome_1 74713 76735 chromosome_1 . 3prime_UTR 73736 76735 . - . ID=evm.TU.chromosome_1.6;Name=EVM%20prediction%20chromosome_1.6
chromosome_1 74713 76735 chromosome_1 . 3prime_UTR 74714 77713 . + . ID=evm.TU.chromosome_1.5;Name=EVM%20prediction%20chromosome_1.5
chromosome_1 74713 76735 chromosome_1 . 3prime_UTR 73736 76735 . - . ID=evm.TU.chromosome_1.6;Name=EVM%20prediction%20chromosome_1.6
The numbers don’t look correct. It’s trying to find the intersections and it is finding them but its the same info because its the 3UTR 3kb downstream gff. There is an associated ID which corresponds to gene but I am hesitant to use it, especially since there are so many rows, there are probably repeats. I could use bed closest, which searches for the nearest feature in the other file. In the scripts folder: nano bed_close_3UTR.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia_Genome/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Astrangia_Genome
echo "Finding closest gene to 3'UTR seqs " $(date)
bedtools closest -a apoc_3UTR.bed -b apoc_GFFannotation.gene_sorted.gff > closest_genes.txt
echo "Complete!" $(date)
Submitted batch job 310687. Got this error:
Error: Sorted input specified, but the file apoc_3UTR.bed has the following out of order record
chromosome_1 92518 93919
Adding this line: sort -k1,1 -k2,2n -o apoc_3UTR_sorted.bed apoc_3UTR.bed to the script. Submitted batch job 310688. Now got this error:
ERROR: chromomsome sort ordering for file apoc_GFFannotation.gene_sorted.gff is inconsistent with other files. Record was:
chromosome_10 . gene 12623 13741 . + . ID=evm.TU.chromosome_10.1;Name=EVM%20prediction%20chromosome_10.1
But an output file did get produced!
wc -l closest_genes.txt
73170 closest_genes.txt
chromosome_1 17663 20663 chromosome_1 EVM gene 20664 21393 . - . ID=evm.TU.chromosome_1.1;Name=EVM%20prediction%20chromosome_1.1
chromosome_1 40489 43489 chromosome_1 . gene 34636 40489 . + . ID=evm.TU.chromosome_1.2;Name=EVM%20prediction%20chromosome_1.2
chromosome_1 53463 55801 chromosome_1 . gene 43758 53463 . + . ID=evm.TU.chromosome_1.3;Name=EVM%20prediction%20chromosome_1.3
chromosome_1 53463 55801 chromosome_1 . gene 55802 60431 . - . ID=evm.TU.chromosome_1.4;Name=EVM%20prediction%20chromosome_1.4
chromosome_1 53463 55801 chromosome_1 . gene 43758 53463 . + . ID=evm.TU.chromosome_1.3;Name=EVM%20prediction%20chromosome_1.3
chromosome_1 53463 55801 chromosome_1 . gene 55802 60431 . - . ID=evm.TU.chromosome_1.4;Name=EVM%20prediction%20chromosome_1.4
chromosome_1 74713 76735 chromosome_1 . gene 62282 74713 . + . ID=evm.TU.chromosome_1.5;Name=EVM%20prediction%20chromosome_1.5
chromosome_1 74713 76735 chromosome_1 . gene 76736 78519 . - . ID=evm.TU.chromosome_1.6;Name=EVM%20prediction%20chromosome_1.6
chromosome_1 74713 76735 chromosome_1 . gene 62282 74713 . + . ID=evm.TU.chromosome_1.5;Name=EVM%20prediction%20chromosome_1.5
chromosome_1 74713 76735 chromosome_1 . gene 76736 78519 . - . ID=evm.TU.chromosome_1.6;Name=EVM%20prediction%20chromosome_1.6
Interesting, there are duplicates, which makes sense since it was doing it for all the genes. I think I need to select the ones on the - strand.
awk '$10 == "-" {print}' closest_genes.txt > closest_genes_neg_strand.txt
wc -l closest_genes_neg_strand.txt
21183 closest_genes_neg_strand.txt
Maybe not? Idk. Some chromosome info is just on the + strand and some on the - strand. An example:
chromosome_1 1315574 1318574 chromosome_1 EVM gene 1318575 1320328 . - . ID=evm.TU.chromosome_1.94;Name=EVM%20prediction%20chromosome_1.94
chromosome_1 1320328 1321584 chromosome_1 EVM gene 1318575 1320328 . - . ID=evm.TU.chromosome_1.94;Name=EVM%20prediction%20chromosome_1.94
chromosome_1 1335009 1336339 chromosome_1 . gene 1330359 1335009 . + . ID=evm.TU.chromosome_1.96;Name=EVM%20prediction%20chromosome_1.96
chromosome_1 1335009 1336339 chromosome_1 . gene 1330359 1335009 . + . ID=evm.TU.chromosome_1.96;Name=EVM%20prediction%20chromosome_1.96
evm.TU.chromosome_1.94 is only on the - strand, even though it shows up twice. evm.TU.chromosome_1.96 is only on the + strand, even though it shows up twice as well. Lot of duplicates? I am confused about the strandness.
Next steps:
- subset
closest_genes.txtto only contain DEGs. - extract 3’UTR sequence info about the DEGs
- Use this info to subset the 3’UTR DEG sequences
20240410
I’m going to subset the closest_genes.txt file to contain only DEGs. This is a list of my differentially expressed genes. I’m copying it to the server into /data/putnamlab/jillashey/Astrangia_Genome. In the closest_genes.txt file, the 12th column has the information about the corresponding gene. This info is under the ID=XXXXX part of the file.
grep -Ff DEG_list.txt closest_genes.txt > deg_closest_genes.txt
wc -l deg_closest_genes.txt
3187 deg_closest_genes.txt
Sanity checking by randomly taking IDs and searching the DEG list. Not seeing all of the DEGs…Ah because grep was looking for anything that matched. So for example, if I had DEG evm.TU.chromosome_4.153, grep would also pull evm.TU.chromosome_4.1535 because it contains the same string. Remove ID= and other extraneous info from last column.
awk 'BEGIN{FS=OFS="\t"} {split($NF, id, ";"); split(id[1], id_value, "="); $NF=id_value[2]; print $0, id_value[1]}' closest_genes.txt > modified_closest_genes.txt
head modified_closest_genes.txt
chromosome_1 17663 20663 chromosome_1 EVM gene 20664 21393 . - . evm.TU.chromosome_1.1 ID
chromosome_1 40489 43489 chromosome_1 . gene 34636 40489 . + . evm.TU.chromosome_1.2 ID
chromosome_1 53463 55801 chromosome_1 . gene 43758 53463 . + . evm.TU.chromosome_1.3 ID
chromosome_1 53463 55801 chromosome_1 . gene 55802 60431 . - . evm.TU.chromosome_1.4 ID
chromosome_1 53463 55801 chromosome_1 . gene 43758 53463 . + . evm.TU.chromosome_1.3 ID
chromosome_1 53463 55801 chromosome_1 . gene 55802 60431 . - . evm.TU.chromosome_1.4 ID
chromosome_1 74713 76735 chromosome_1 . gene 62282 74713 . + . evm.TU.chromosome_1.5 ID
chromosome_1 74713 76735 chromosome_1 . gene 76736 78519 . - . evm.TU.chromosome_1.6 ID
chromosome_1 74713 76735 chromosome_1 . gene 62282 74713 . + . evm.TU.chromosome_1.5 ID
chromosome_1 74713 76735 chromosome_1 . gene 76736 78519 . - . evm.TU.chromosome_1.6 ID
Not sure why it make an column that just has “ID” in it but whatever. Count number of columns and rows:
awk '{print NF}' modified_closest_genes.txt | sort -nu | tail -n 1
# 13 columns
wc -l modified_closest_genes.txt
# 73170 rows
Remove any duplicate rows
awk '!seen[$0]++' modified_closest_genes.txt > uniq_modified_closest_genes.txt
awk '{print NF}' uniq_modified_closest_genes.txt | sort -nu | tail -n 1
# 13 columns
wc -l uniq_modified_closest_genes.txt
57300 uniq_modified_closest_genes.txt
Okay now I can filter uniq_modified_closest_genes.txt by the DEG list.
awk 'NR==FNR{deg[$1]; next} $12 in deg' DEG_list.txt uniq_modified_closest_genes.txt > uniq_modified_closest_degs.txt
head uniq_modified_closest_degs.txt
chromosome_1 987846 990846 chromosome_1 . gene 990847 993879 . - . evm.TU.chromosome_1.74 ID
chromosome_1 993879 996650 chromosome_1 . gene 990847 993879 . - . evm.TU.chromosome_1.74 ID
chromosome_1 1335009 1336339 chromosome_1 . gene 1330359 1335009 . + . evm.TU.chromosome_1.96 ID
chromosome_1 1397052 1400052 chromosome_1 EVM gene 1400053 1410554 . - . evm.TU.chromosome_1.105 ID
chromosome_1 2741797 2744797 chromosome_1 . gene 2744798 2746546 . - . evm.TU.chromosome_1.220 ID
chromosome_1 3378499 3381499 chromosome_1 . gene 3381500 3384205 . - . evm.TU.chromosome_1.267 ID
chromosome_1 3387647 3390647 chromosome_1 . gene 3390648 3394579 . - . evm.TU.chromosome_1.268 ID
chromosome_1 3452321 3455321 chromosome_1 . gene 3455322 3508018 . - . evm.TU.chromosome_1.272 ID
chromosome_1 3790853 3791719 chromosome_1 EVM gene 3791720 3796967 . - . evm.TU.chromosome_1.303 ID
chromosome_1 4998359 5000981 chromosome_1 . gene 4997077 4998359 . + . evm.TU.chromosome_1.419 ID
wc -l uniq_modified_closest_degs.txt
956 uniq_modified_closest_degs.txt
Does this make sense? That not all of the DEGs are represented in the 3’UTRs? I also see there are some duplicates (evm.TU.chromosome_1.74) where two 3’UTR sequences were closest to evm.TU.chromosome_1.74 coordinates.
tail uniq_modified_closest_degs.txt
chromosome_9 23374102 23375784 chromosome_9 . gene 23369259 23374102 . - . evm.TU.chromosome_9.2218 ID
chromosome_9 23399610 23402610 chromosome_9 . gene 23402611 23417409 . - . evm.TU.chromosome_9.2221 ID
chromosome_9 23472207 23473119 chromosome_9 . gene 23469848 23472207 . + . evm.TU.chromosome_9.2232 ID
chromosome_9 23484687 23486331 chromosome_9 . gene 23486332 23493309 . - . evm.TU.chromosome_9.2235 ID
chromosome_9 23643484 23646484 chromosome_9 . gene 23646485 23658996 . - . evm.TU.chromosome_9.2248 ID
chromosome_9 23662284 23664931 chromosome_9 EVM gene 23664932 23667853 . + . evm.TU.chromosome_9.2250 ID
chromosome_9 23667853 23669741 chromosome_9 EVM gene 23664932 23667853 . + . evm.TU.chromosome_9.2250 ID
chromosome_9 23837944 23840944 chromosome_9 . gene 23832305 23837944 . + . evm.TU.chromosome_9.2269 ID
chromosome_9 24085331 24088331 chromosome_9 . gene 24088332 24106631 . - . evm.TU.chromosome_9.2293 ID
chromosome_9 24157356 24160356 chromosome_9 . gene 24160357 24173706 . - . evm.TU.chromosome_9.2299 ID
Only goes up to chromosome 9?? Weird. Even closest_genes.txt file only goes up to chromosome 9…I think it goes back to this error from above:
ERROR: chromomsome sort ordering for file apoc_GFFannotation.gene_sorted.gff is inconsistent with other files. Record was:
chromosome_10 . gene 12623 13741 . + . ID=evm.TU.chromosome_10.1;Name=EVM%20prediction%20chromosome_10.1
Let’s try to resort the gff?
sort -k1,1 -k4,4n apoc_GFFannotation.gene_sorted.gff > sorted_apoc_GFFannotation.gene_sorted.gff
Ah so its sorting it so chromosome 1 is first, then 10, etc. 9 is last because it is that last number in 1-9. But the original file (apoc_GFFannotation.gene_sorted.gff) is sorted by chromosome? I am so frustrated haha. Look at all lines with chromosome 10:
awk '$1 == "chromosome_10"' apoc_GFFannotation.gene_sorted.gff
Weird, it looks like chromosome 10 is repeating itself?
chromosome_10 EVM gene 33840711 33841013 . - . ID=evm.TU.chromosome_10.3274;Name=EVM%20prediction%20chromosome_10.3274
chromosome_10 . gene 33859427 33860415 . + . ID=evm.TU.chromosome_10.3275;Name=EVM%20prediction%20chromosome_10.3275
chromosome_10 . gene 12623 13741 . + . ID=evm.TU.chromosome_10.1;Name=EVM%20prediction%20chromosome_10.1
chromosome_10 . gene 15707 16786 . - . ID=evm.TU.chromosome_10.2;Name=EVM%20prediction%20chromosome_10.2
chromosome_10 EVM gene 18626 18934 . + . ID=evm.TU.chromosome_10.3;Name=EVM%20prediction%20chromosome_10.3
chromosome_10 . gene 22435 26984 . - . ID=evm.TU.chromosome_10.4;Name=EVM%20prediction%20chromosome_10.4
chromosome_10 EVM gene 29369 30803 . + . ID=evm.TU.chromosome_10.5;Name=EVM%20prediction%20chromosome_10.5
chromosome_10 EVM gene 31529 36887 . - . ID=evm.TU.chromosome_10.6;Name=EVM%20prediction%20chromosome_10.6
It ends with evm.TU.chromosome_10.3275, then restarts with evm.TU.chromosome_10.1…Are all of the chromosomes 10-14 like this?
awk '$1 == "chromosome_11"' apoc_GFFannotation.gene_sorted.gff
Nope 11 doesn’t look like that. Let’s try to remove duplicate rows from apoc_GFFannotation.gene_sorted.gff?
wc -l apoc_GFFannotation.gene_sorted.gff
47156 apoc_GFFannotation.gene_sorted.gff
awk '!seen[$0]++' apoc_GFFannotation.gene_sorted.gff > uniq_apoc_GFFannotation.gene_sorted.gff
wc -l uniq_apoc_GFFannotation.gene_sorted.gff
47156 uniq_apoc_GFFannotation.gene_sorted.gff
This didn’t do anything. Looking at the bed_close_3UTR.sh script, I have a line that does: sort -k1,1 -k2,2n -o apoc_3UTR_sorted.bed apoc_3UTR.bed aka not sorting by bedtools. In this line: bedtools closest -a apoc_3UTR_sorted.bed -b apoc_GFFannotation.gene_sorted.gff > closest_genes.txt, let’s try to sub apoc_GFFannotation.gene_sorted.gff for sorted_apoc_GFFannotation.gene_sorted.gff. Submitted batch job 311017. No error message produced.
wc -l closest_genes.txt
89784 closest_genes.txt
Now let’s try this again. Make Id column.
awk 'BEGIN{FS=OFS="\t"} {split($NF, id, ";"); split(id[1], id_value, "="); $NF=id_value[2]; print $0, id_value[1]}' closest_genes.txt > modified_closest_genes.txt
wc -l modified_closest_genes.txt
89784 modified_closest_genes.txt
Remove any duplicate rows
awk '!seen[$0]++' modified_closest_genes.txt > uniq_modified_closest_genes.txt
wc -l uniq_modified_closest_genes.txt
67788 uniq_modified_closest_genes.txt
Okay now I can filter uniq_modified_closest_genes.txt by the DEG list.
awk 'NR==FNR{deg[$1]; next} $12 in deg' DEG_list.txt uniq_modified_closest_genes.txt > uniq_modified_closest_degs.txt
Sanity check! Check some of the IDs from uniq_modified_closest_degs.txt against the DEG list. Okay looking good so far. Now I need to make a new column that combines columns 1, 2, and 3 (ie the 3UTR info columns) so that a new column is created that it represents the file headers in apoc_3UTR.fasta. Ie the new column should look like this: chromosome_1:17663-20663.
awk '{print $1":"$2"-"$3, $0}' uniq_modified_closest_degs.txt > uniq_modified_closest_degs_3UTRid.txt
head uniq_modified_closest_degs_3UTRid.txt
chromosome_1:987846-990846 chromosome_1 987846 990846 chromosome_1 . gene 990847 993879 . - . evm.TU.chromosome_1.74 ID
chromosome_1:993879-996650 chromosome_1 993879 996650 chromosome_1 . gene 990847 993879 . - . evm.TU.chromosome_1.74 ID
chromosome_1:1335009-1336339 chromosome_1 1335009 1336339 chromosome_1 . gene 1330359 1335009 . + . evm.TU.chromosome_1.96 ID
chromosome_1:1397052-1400052 chromosome_1 1397052 1400052 chromosome_1 EVM gene 1400053 1410554 . - . evm.TU.chromosome_1.105 ID
chromosome_1:2741797-2744797 chromosome_1 2741797 2744797 chromosome_1 . gene 2744798 2746546 . - . evm.TU.chromosome_1.220 ID
chromosome_1:3378499-3381499 chromosome_1 3378499 3381499 chromosome_1 . gene 3381500 3384205 . - . evm.TU.chromosome_1.267 ID
chromosome_1:3387647-3390647 chromosome_1 3387647 3390647 chromosome_1 . gene 3390648 3394579 . - . evm.TU.chromosome_1.268 ID
chromosome_1:3452321-3455321 chromosome_1 3452321 3455321 chromosome_1 . gene 3455322 3508018 . - . evm.TU.chromosome_1.272 ID
chromosome_1:3790853-3791719 chromosome_1 3790853 3791719 chromosome_1 EVM gene 3791720 3796967 . - . evm.TU.chromosome_1.303 ID
chromosome_1:4998359-5000981 chromosome_1 4998359 5000981 chromosome_1 . gene 4997077 4998359 . + . evm.TU.chromosome_1.419 ID
Separate the new column into its own text file.
awk '{print $1}' uniq_modified_closest_degs_3UTRid.txt > DEG_3UTR.txt
Now subset the 3’UTR fasta file based on the 3’UTR DEG seqs in the DEG_3UTR.txt list.
grep -A 1 -f DEG_3UTR.txt apoc_3UTR.fasta | grep -v "^--$" > 3UTR_DE.fa
zgrep -c ">" 3UTR_DE.fa
2179
Important note: uniq_modified_closest_degs_3UTRid.txt has the DEG and 3’UTR ids in it. I copied this file, along with 3UTR_DE.fa to my local computer.
I believe I can run miranda now!!!!!!!!! In /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts: nano miranda_de.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "starting miranda run with differentially expressed genes and miRNAs with score cutoff >100 and energy cutoff <-10"$(date)
module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_DE.fa /data/putnamlab/jillashey/Astrangia_Genome/3UTR_DE.fa -sc 100 -en -10 -out /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_de.tab
conda deactivate
echo "miranda run finished!"$(date)
echo "counting number of putative interactions predicted"$(date)
zgrep -c "Performing Scan" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_de.tab
Submitted batch job 311424
20240411
Miranda finished running in about 15 mins and ~108,000 putative interactions were predicted! This is great! Let’s look at some of the results:
Forward: Score: 123.000000 Q:2 to 18 R:676 to 694 Align Len (16) (81.25%) (87.50%)
Query: 3' cccTTGTTCGGCTTTGTAAc 5'
|||||||| |||: ||
Ref: 5' tcaAACAAGCC-AAATTTTc 3'
Energy: -12.090000 kCal/Mol
Scores for this hit:
>chromosome_12_481048 chromosome_1:987846-990846 123.00 -12.09 2 18 676 694 16 81.25% 87.50%
Woohoo. In this example, there isn’t exactly strict seed binding and there is a G:U wobble pair. Once again, how do I invoke strict seed binding?? A comment on this biostars question about miranda output meaning said: “No, I’m currently still using miRanda but I might complement it by using other Tools. Anyway, if you do not use the default parameters and therefore add this function “ -strict “ in the command, this means that you require strict alignment in the seed region (position 2-8). We already identified some interesting targets.” In the manual, it does not say anything about -strict function. Maybe I’ll try it? Added the strict flag and renamed the output file. Submitted batch job 311700. The question also recommended the following to parse the miranda output:
grep -A 1 "Scores for this hit:" miranda_out.txt | sort | grep '>'
Which will provide me with this:
>mirna_name transcript_target 143.00 -22.87 2 18 339 357 16 75.00% 87.50%
With the header being this:
mirna Target Score Energy-Kcal/Mol Query-Aln(start-end) Subjetct-Al(Start-End) Al-Len Subject-Identity Query-Identity
The strict miranda ran, but no difference in number of outputs it looks like. But there looks to be less info in the strict file:
ls -othr
total 355M
-rw-r--r--. 1 jillashey 315M Apr 10 11:57 miranda_de.tab
-rw-r--r--. 1 jillashey 40M Apr 11 10:16 miranda_de_strict.tab
Let’s parse the outputs from both files.
grep -A 1 "Scores for this hit:" miranda_de.tab | sort | grep '>' > miranda_de_parsed.txt
wc -l miranda_de_parsed.txt
750674 miranda_de_parsed.txt
grep -A 1 "Scores for this hit:" miranda_de_strict.tab | sort | grep '>' > miranda_de_strict_parsed.txt
wc -l miranda_de_strict_parsed.txt
14899 miranda_de_strict_parsed.txt
Strict definitely resulted in less hits, which is probably good because I want the binding to be pretty stringent, given the strict seed binding that appears to be present in cnidarians. Yay!!!!!!! Copying miranda_de_strict_parsed.txt to my local computer.
20240421
I’m going to now run miranda on all miRNAs and mRNAs.
In /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts: nano miranda_strict_all.sh
#!/bin/bash -i
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB --cpus-per-task=24
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "starting miranda run with all genes and miRNAs with score cutoff >100, energy cutoff <-10, and strict binding invoked"$(date)
module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa /data/putnamlab/jillashey/Astrangia_Genome/apoc_3UTR.fasta -sc 100 -en -10 -strict -out /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all.tab
conda deactivate
echo "miranda run finished!"$(date)
echo "counting number of putative interactions predicted" $(date)
zgrep -c "Performing Scan" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all.tab
echo "Parsing output" $(date)
grep -A 1 "Scores for this hit:" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all.tab | sort | grep '>' > /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_parsed.txt
echo "counting number of putative interactions predicted" $(date)
wc -l /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_parsed.txt
echo "miranda script complete" $(date)
Submitted batch job 312601. Job might pend for a while because it requires a lot of resources. Started running after about 13 hours.
20240422
Now that the job above is running, I’m going to run miranda with the miRNAs and the lncRNAs. lncRNAs may bind to miRNAs and “sponge” them up, essentially sequestering them so that they can’t bind to their target mRNAs and degrade those mRNAs.
In /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts: nano miranda_strict_lncRNA.sh
#!/bin/bash -i
#SBATCH -t 200:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB --cpus-per-task=24
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "starting miranda run with all lncRNAs and miRNAs with score cutoff >100, energy cutoff <-10, and strict binding invoked"$(date)
module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/all/mirna_results_04_02_2024_t_11_15_57/mature_all.fa /data/putnamlab/jillashey/Astrangia2021/lncRNA/output/CPC2/apoc_bedtools_lncRNAs.fasta -sc 100 -en -10 -strict -out /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_lncRNA.tab
conda deactivate
echo "miranda run finished!"$(date)
echo "counting number of putative interactions predicted" $(date)
zgrep -c "Performing Scan" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_lncRNA.tab
echo "Parsing output" $(date)
grep -A 1 "Scores for this hit:" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_lncRNA.tab | sort | grep '>' > /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_lncRNA_parsed.txt
echo "counting number of putative interactions predicted" $(date)
wc -l /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_lncRNA_parsed.txt
echo "miranda script complete" $(date)
Submitted batch job 312886
20240426
Both miranda jobs have preemptively failed and restarted. Not sure why…Going back to what Kevin Bryan said to me regarding this in the Apul genome assembly: “you didn’t specify -t SLURM_CPUS_ON_NODE (and also #SBATCH –exclusive) to make use of all of the CPU cores on the node. You might want to consider re-submitting this job with those parameters. Because the nodes generally have 36 cores, it should be able to catch up to where it is now in a little over half a day, assuming perfect scaling.” Looking at the miranda manual, it doesn’t look like there is any flag to specify cpus on node. Cancelled jobs 312601 and 312886. I am going to redo the SLURM info in these scripts. I edited both miranda_strict_all.sh and miranda_strict_lncRNA.sh so that the slurm headers look like:
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
For miranda_strict_all.sh: Submitted batch job 313313. For miranda_strict_lncRNA.sh: Submitted batch job 313314. Both started running right away!
20240517
Over the last few weeks, I downloaded the mRNA miranda results and identified correlations between differentially expressed mRNAs and miRNAs (see code here). I have now isolated 51 genes that are shared across 4 comparisons (TP0vTP5 amb, TP0vTP7amb, TP0vTP5 heat, and TP0vTP7 heat) (see csv of that list hereXXXXX). With this list, I want to subset those genes from the mRNA fasta and blast them against the protein db.
I also might run interproscan…idk yet.
cd /data/putnamlab/jillashey/Astrangia2021/mRNA
mkdir blast
cd blast
Copied in sigcorr_subset.fasta, which I made on my computer. In the scripts folder: nano sigcorr_blast.sh
#!/bin/bash
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=125GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/mRNA/blast
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
cd /data/putnamlab/jillashey/Astrangia2021/mRNA/blast
echo "Blasting Apoc genes of interest (significant correlation with miRNAs) against remote nt database" $(date)
blastn -query sigcorr_subset.fasta -db nt -evalue 1E-40 -num_threads 10 -max_target_seqs 3 -max_hsps 3 -outfmt 6 -out sigcorr_subset_blast_results.txt
echo "Blast complete" $(date)
Submitted batch job 317026. Also going to submit a blastx job. Submitted batch job 317071
20240812
Zoe found this great tool called GeneExt, which is used to adjust/extend genes so that the 3’UTRs are annotated based on the mapping of reads, helping with overall mapping in scRNA and tag-seq. I’m going to use it to find the 3’UTRs for my genes! Cnidarian miRNAs bind to the 3’UTR of genes and in the code above, I estimated that the 3’UTRs are 3kb before the gene. However, this was just an estimate and I want to use GeneExt to find the 3’UTRs in a quantified way. I’m using Zoe’s code.
First, convert gff3 to gtf using gffread. In the /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts scripts folder: nano gffread.sh
#!/bin/bash
#SBATCH -t 120:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --mem=250GB
#SBATCH --account=putnamlab
#SBATCH --export=NONE
#SBATCH --error="%x_error.%j" #if your job fails, the error report will be put in this file
#SBATCH --output="%x_output.%j" #once your job is completed, any final job report comments will be put in this file
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
# load modules needed
module load gffread/0.12.7-GCCcore-11.2.0
cd /data/putnamlab/jillashey/Astrangia_Genome/
# Combined code, want to see if these are any different
gffread -E apoculata_v2.0.gff3 -T -o apoculata_v2.0.gtf
Submitted batch job 334287. Ran super fast. Checked gtf file, looks good. Next, combine 20 bam files together from the mRNA mapping step.
In the /data/putnamlab/jillashey/Astrangia2021/mRNA/scripts folder, nano merge_bam.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=125GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/mRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load SAMtools/1.9-foss-2018b
cd /data/putnamlab/jillashey/Astrangia2021/mRNA/output/bowtie/align
#use samtools merge to merge all the files
samtools merge AST_merge.bam *.bam
Submitted batch job 334289. While this is running, append apoculata_v2.0_modified.gtf in /data/putnamlab/jillashey/Astrangia_Genome so that the transcript_ids have a -T at the end. The gene and transcript ids in the gtf must be different for GeneExt.
cd /data/putnamlab/jillashey/Astrangia_Genome
sed 's/transcript_id "\([^"]*\)"/transcript_id "\1-T"/g' apoculata_v2.0.gtf > apoculata_v2.0_modified.gtf
Zoe already loaded the GeneExt program into her directory here: /data/putnamlab/zdellaert/snRNA/programs/GeneExt but I couldn’t get it to activate so I’m going to try to install it here /data/putnamlab/conda.
cd /data/putnamlab/conda
mkdir GeneExt
cd GeneExt
git clone https://github.com/sebepedroslab/GeneExt.git
interactive
module load Miniconda3/4.9.2
conda env create -n geneext -f environment.yaml
Probably going to run for a while. Once this is installed, I can run GeneExt!
20240813
Installation for GeneExt ran for most of the day yesterday, then quit. Updating it now:
cd /data/putnamlab/conda/GeneExt/
interactive -c 10
module load Miniconda3/4.9.2
conda env update -n geneext -f environment.yaml
This ran for about an hour but successfully updated. Let’s try to run the test data to make sure the program works.
exit # exit previous interactive mode
interactive -c 10
conda activate geneext
# test run
python geneext.py -g test_data/annotation.gtf -b test_data/alignments.bam -o result.gtf --peak_perc 0
Output:
____ _____ _
/ ___| ___ _ __ ___| ____|_ _| |_
| | _ / _ \ '_ \ / _ \ _| \ \/ / __|
| |_| | __/ | | | __/ |___ > <| |_
\____|\___|_| |_|\___|_____/_/\_\__|
______ ___ ______
-----[______]==[___]==[______]===>----
Gene model adjustment for improved single-cell RNA-seq data counting
╭──────────────────╮
│ Preflight checks │
╰──────────────────╯
Genome annotation warning: Could not find "gene" features in test_data/annotation.gtf! Trying to fix ...
╭───────────╮
│ Execution │
╰───────────╯
Running macs2 ...
########## macs2 FAILED ##############
return code: 1
Output: Traceback (most recent call last):
File "/home/jillashey/.conda/envs/geneext/bin/macs2", line 653, in <module>
main()
File "/home/jillashey/.conda/envs/geneext/bin/macs2", line 49, in main
from MACS2.callpeak_cmd import run
File "/home/jillashey/.conda/envs/geneext/lib/python3.9/site-packages/MACS2/callpeak_cmd.py", line 23, in <module>
from MACS2.OptValidator import opt_validate
File "/home/jillashey/.conda/envs/geneext/lib/python3.9/site-packages/MACS2/OptValidator.py", line 20, in <module>
from MACS2.IO.Parser import BEDParser, ELANDResultParser, ELANDMultiParser, \
File "__init__.pxd", line 206, in init MACS2.IO.Parser
ValueError: numpy.dtype size changed, may indicate binary incompatibility. Expected 96 from C header, got 88 from PyObject
Traceback (most recent call last):
File "/glfs/brick01/gv0/putnamlab/conda/GeneExt/geneext.py", line 703, in <module>
helper.run_macs2(tempdir+'/' + 'plus.bam','plus',tempdir,verbose = verbose)
File "/glfs/brick01/gv0/putnamlab/conda/GeneExt/geneext/helper.py", line 79, in run_macs2
ps.check_returncode()
File "/home/jillashey/.conda/envs/geneext/lib/python3.9/subprocess.py", line 460, in check_returncode
raise CalledProcessError(self.returncode, self.args, self.stdout,
subprocess.CalledProcessError: Command '('macs2', 'callpeak', '-t', 'tmp/plus.bam', '-f', 'BAM', '--keep-dup', '20', '-q', '0.01', '--shift', '1', '--extsize', '100', '--broad', '--nomodel', '--min-length', '30', '-n', 'plus', '--outdir', 'tmp')' returned non-zero exit status 1.
Something went wrong.
20240819
GeneExt is being weird when installing so I’m going to delete what I have so far and restart. BLEH.
interactive
module load Miniconda3/4.9.2
cd /data/putnamlab/conda
rm -r GeneExt/
git clone https://github.com/sebepedroslab/GeneExt.git
cd GeneExt/
conda env create -n geneext -f environment.yaml
This took much faster than last time…Let’s try to run the test data?
exit # need to exit interactive mode after I create the env
interactive
conda activate geneext
# test run
python geneext.py -g test_data/annotation.gtf -b test_data/alignments.bam -o result.gtf --peak_perc 0
Got same error as above. I hate myself. Going to email Kevin Bryan. Okay while I go insane with this, I want to run ShortStack on my AST data. We ended up using this program for the e5 deep dive ncRNA paper, it was recommended by Javi, who has used it in the past.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA
mkdir shortstack
Kevin Bryan already installed short stack on Andromeda (thank god). Trimmed reads are in /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar. Sam merged R1 and R2 before running. I am going to just run on R1 for a first pass. In the scripts folder: nano shortstack_test.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Running short stack on mature trimmed miRNAs (R1) from AST project"
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
# Load modules
module load ShortStack/4.0.2-foss-2022a
# Run short stack
ShortStack \
--genomefile /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta \
--readfile AST-1065_R1_001.fastq.gz_1.fastq \
AST-1147_R1_001.fastq.gz_1.fastq \
AST-1412_R1_001.fastq.gz_1.fastq \
AST-1560_R1_001.fastq.gz_1.fastq \
AST-1567_R1_001.fastq.gz_1.fastq \
AST-1617_R1_001.fastq.gz_1.fastq \
AST-1722_R1_001.fastq.gz_1.fastq \
AST-2000_R1_001.fastq.gz_1.fastq \
AST-2007_R1_001.fastq.gz_1.fastq \
AST-2302_R1_001.fastq.gz_1.fastq \
AST-2360_R1_001.fastq.gz_1.fastq \
AST-2398_R1_001.fastq.gz_1.fastq \
AST-2404_R1_001.fastq.gz_1.fastq \
AST-2412_R1_001.fastq.gz_1.fastq \
AST-2512_R1_001.fastq.gz_1.fastq \
AST-2523_R1_001.fastq.gz_1.fastq \
AST-2563_R1_001.fastq.gz_1.fastq \
AST-2729_R1_001.fastq.gz_1.fastq \
AST-2755_R1_001.fastq.gz_1.fastq \
--known_miRNAs /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa \
--outdir /data/putnamlab/jillashey/Astrangia2021/smRNA/shortstack \
--threads 10 \
--dn_mirna
echo "Short stack run complete"
Submitted batch job 334980. Failed with this error: Required executable wigToBigWig : Not found!. Confused by this…apparently wigToBigWig is a type of file format. Not sure what to do with this info…Going to email Kevin Bryan!
20240829
Kevin Bryan got back to me and said to add the module Kent_tools/442-GCC-11.3.0 to find wigToBigWig for the short stack code. Submitted batch job 336294. Running successfully!
I also emailed him about the issues installing gene ext and he said “it looks like you have two different genext environments loaded there since some paths refer to /home/jillashey/.conda/envs/geneext and others to /glfs/brick01/gv0/putnamlab/conda/GeneExt, so I think that’s what’s causing the issue. You might want to delete both and recreate the second one to ensure it has all the dependencies.” I deleted gene ext in /home/jillashey/.conda/envs/ directory. Went to /glfs/brick01/gv0/putnamlab/conda/ and deleted gene ext. Because it takes a while for gene ext to install, I’m going to run the get_geneext.sh script. Submitted batch job 336304.
20240830
Gene ext installed but once again, got the same error message even though I thought I deleted it from all other locations. Here’s the error once again:
########## macs2 FAILED ##############
return code: 1
Output: Traceback (most recent call last):
File "/home/jillashey/.conda/envs/geneext/bin/macs2", line 653, in <module>
main()
File "/home/jillashey/.conda/envs/geneext/bin/macs2", line 49, in main
from MACS2.callpeak_cmd import run
File "/home/jillashey/.conda/envs/geneext/lib/python3.9/site-packages/MACS2/callpeak_cmd.py", line 23, in <module>
from MACS2.OptValidator import opt_validate
File "/home/jillashey/.conda/envs/geneext/lib/python3.9/site-packages/MACS2/OptValidator.py", line 20, in <module>
from MACS2.IO.Parser import BEDParser, ELANDResultParser, ELANDMultiParser, \
File "__init__.pxd", line 206, in init MACS2.IO.Parser
ValueError: numpy.dtype size changed, may indicate binary incompatibility. Expected 96 from C header, got 88 from PyObject
Maybe it’s something in here: /home/jillashey/.conda/pkgs but there are so many folders and tools in there that I am not sure which one to delete. But shortstack did run! Look at the out file in the scripts folder (slurm-336294.out):
Found a total of 51 MIRNA loci
Writing final files
Non-MIRNA loci by DicerCall:
N 9093
23 48
22 38
21 16
24 12
Creating visualizations of microRNA loci with strucVis
<<< WARNING >>>
Do not rely on these results alone to annotate new MIRNA loci!
The false positive rate for de novo MIRNA identification is low, but NOT ZERO
Insepct each mirna locus, especially the strucVis output, and see
https://doi.org/10.1105/tpc.17.00851 , https://doi.org/10.1093/nar/gky1141
Thu 29 Aug 2024 19:06:45 -0400 EDT
Run Completed!
Short stack run complete
The out file found 51 putative miRNAs.
Look at shortstack results (using e5 deep dive shortstack code as a reference).
echo "Number of potential loci:"
awk '(NR>1)' Results.txt | wc -l
Nummber of potential loci:
9258
Column 20 in the Results.txt file identifies if a cluster is an miRNA or not (Y or N). 51 loci are characterized as miRNA and 9207 loci are not characterized as miRNAs. Column 21 in the Results.txt file identifies if a cluster aligned to a known miRNA or not (Y or N). 37 loci aligned to a known miRNA and 9222 loci did not. However, there are only 5 valid miRNAs that match up with known miRNAs
column: line 3 is too long, output will be truncated
chromosome_6:21902734-21902826 Y nve-miR-9425_MIMAT0035384_Nematostella_vectensis_miR-9425;miR-9425_Nematostella_vectensis_Moran_et_al._2014_NA
chromosome_7:2303817-2303911 Y sca-nve-F-miR-2036_Scolanthus_callimorphus_Praher_et_al._2021_Transcriptome-level;eca-nve-F-miR-2036_Edwardsiella_carnea_Praher_et_al._2021_Transcriptome-level
0-5p;ccr-miR-100_MIMAT0026195_Cyprinus_carpio_miR-100;pmi-miR-100-5p_MIMAT0032156_Patiria_miniata_miR-100-5p;cpi-miR-100-5p_MIMAT0037714_Chrysemys_picta_miR-100-5p;chi-miR-100-5p_MIMAT0035897_Capra_hircus_miR-100-5p;dma-miR-100_MIMAT0049252_Daubentonia_madagascariensis_miR-100;sbo-miR-100_MIMAT0049501_Saimiri_boliviensis_miR-100;ola-miR-100_MIMAT0022614_Oryzias_latipes_miR-100
chromosome_8:4117884-4117974 Y Adi-Mir-2030_5p_Acropora_digitifera_Gajigan_&_Conaco_2017_nve-miR-2030-5p;_nve-miR-2030-5p;_spi-miR-temp-40;ami-nve-F-miR-2030-5p_Acropora_millepora_Praher_et_al._2021_NA;adi-nve-F-miR-2030_Acropora_digitifera__Praher_et_al._2021_NA;spi-mir-temp-40_Stylophora_pistillata_Liew_et_al._2014_Close_match_of_nve-miR-2030;eca-nve-F-miR-2030_Edwardsiella_carnea_Praher_et_al._2021_Transcriptome-level;miR-2030_Nematostella_vectensis_Moran_et_al._2014_NA
chromosome_14:8601339-8601434 Y apa-mir-2037_Exaiptasia_pallida_Baumgarten_et_al._2017_miR-2037;_Nve;_Spis;spi-mir-temp-20_Stylophora_pistillata_Liew_et_al._2014_NA;eca-nve-F-miR-2037_Edwardsiella_carnea_Praher_et_al._2021_Transcriptome-level;spi-nve-F-miR-2037_Stylophora_pistillata_Praher_et_al._2021_NA;sca-nve-F-miR-2037-3p_Scolanthus_callimorphus_Praher_et_al._2021_Transcriptome-level;ami-nve-F-miR-2037-3p_Acropora_millepora_Praher_et_al._2021_NA;mse-nve-F-miR-2037-3p_Metridium_senile_Praher_et_al._2021_Transcriptome-level;avi-miR-temp-2037_Anemonia_viridis_Urbarova_et_al._2018_NA
Sam White found a bug in the shortstack code that was making the Results.gff3 starting coordinates 1 greater than those listed in the FastA description lines. For example:
grep "Cluster_1155.mature" Results.gff3 mir.fasta
Results.gff3:chromosome_5 ShortStack mature_miRNA 8149619 8149640 822 - . ID=Cluster_1155.mature;Parent=Cluster_1155
mir.fasta:>Cluster_1155.mature::chromosome_5:8149618-8149640(-)
The Results.gff3 file has Cluster_1155 starting at position 8149619, while mir.fasta has it starting at position 8149618. This is an issue because the shortstack fasta headers and gff information need to match or there will be downstream issues. I will need to clean up the fasta with Sam’s code.
20240901
Attempting gene ext installation again.
cd /data/putnamlab/conda
git clone https://github.com/sebepedroslab/GeneExt.git
interactive
module load Miniconda3/4.9.2
cd GeneExt
conda env create --prefix /data/putnamlab/conda/GeneExt/geneext -f environment.yaml
python geneext.py -g test_data/annotation.gtf -b test_data/alignments.bam -o result.gtf --peak_perc 0
____ _____ _
/ ___| ___ _ __ ___| ____|_ _| |_
| | _ / _ \ '_ \ / _ \ _| \ \/ / __|
| |_| | __/ | | | __/ |___ > <| |_
\____|\___|_| |_|\___|_____/_/\_\__|
______ ___ ______
-----[______]==[___]==[______]===>----
Gene model adjustment for improved single-cell RNA-seq data counting
╭──────────────────╮
│ Preflight checks │
╰──────────────────╯
Genome annotation warning: Could not find "gene" features in test_data/annotation.gtf! Trying to fix ...
╭───────────╮
│ Execution │
╰───────────╯
Running macs2 ...
########## macs2 FAILED ##############
return code: 1
Output: Traceback (most recent call last):
File "/data/putnamlab/conda/GeneExt/geneext/bin/macs2", line 653, in <module>
main()
File "/data/putnamlab/conda/GeneExt/geneext/bin/macs2", line 49, in main
from MACS2.callpeak_cmd import run
File "/data/putnamlab/conda/GeneExt/geneext/lib/python3.9/site-packages/MACS2/callpeak_cmd.py", line 23, in <module>
from MACS2.OptValidator import opt_validate
File "/data/putnamlab/conda/GeneExt/geneext/lib/python3.9/site-packages/MACS2/OptValidator.py", line 20, in <module>
from MACS2.IO.Parser import BEDParser, ELANDResultParser, ELANDMultiParser, \
File "__init__.pxd", line 206, in init MACS2.IO.Parser
ValueError: numpy.dtype size changed, may indicate binary incompatibility. Expected 96 from C header, got 88 from PyObject
Traceback (most recent call last):
File "/glfs/brick01/gv0/putnamlab/conda/GeneExt/geneext.py", line 703, in <module>
helper.run_macs2(tempdir+'/' + 'plus.bam','plus',tempdir,verbose = verbose)
File "/glfs/brick01/gv0/putnamlab/conda/GeneExt/geneext/helper.py", line 79, in run_macs2
ps.check_returncode()
File "/data/putnamlab/conda/GeneExt/geneext/lib/python3.9/subprocess.py", line 460, in check_returncode
raise CalledProcessError(self.returncode, self.args, self.stdout,
subprocess.CalledProcessError: Command '('macs2', 'callpeak', '-t', 'tmp/plus.bam', '-f', 'BAM', '--keep-dup', '20', '-q', '0.01', '--shift', '1', '--extsize', '100', '--broad', '--nomodel', '--min-length', '30', '-n', 'plus', '--outdir', 'tmp')' returned non-zero exit status 1.
Still getting same error but now it seems to be in the correct location.
20240912
Kevin Wong is also running into the same error when installing Gene Ext on Umiami server. Zoe did install Gene Ext successfully in June on Andromeda and Unity. Tried activating conda env in her folder on Andromeda: /data/putnamlab/zdellaert/snRNA/programs/GeneExt but do not have permissions. Asked her to run chmod o+rwx /data/putnamlab/zdellaert/snRNA/programs/GeneEx
Let’s see if it works now.
interactive
module load Miniconda3/4.9.2
cd /data/putnamlab/conda
git clone https://github.com/sebepedroslab/GeneExt.git
cd GeneExt/
conda env create -n geneext -f environment.yaml
20241023
Hello world. Stuff still didnt work but Kevin got it to work on the UMiami server!!! He had to update numpy and pandas versions. See his post here.
Going to remove GeneExt and try to reinstall by updating the python package versions.
module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/GeneExt
20241113
Hello again. I decided not to run gene ext – I did it with the e5 data and the estimates of the 3’UTRs were ~1000bp, which was my estimate as well. So I am going to clean up my shortstack output using Sam’s code.
Examine the Results.txt file.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/shortstack
head Results.txt | column -t
Locus Name Chrom Start End Length Reads UniqueReads FracTop Strand MajorRNA MajorRNAReads Short Long 21 22 23 24 DicerCall MIRNA known_miRNAs
chromosome_1:18460-18882 Cluster_1 chromosome_1 18460 18882 423 460 107 0.9108695652173913 + UCAAACCAACGCAGCCGAAAAGUAGAUGU 131 18 414 2 1 15 10 N N NA
chromosome_1:70293-70717 Cluster_2 chromosome_1 70293 70717 425 62964 382 1.0 + UAAGACUCUGAGGAUGGAAU 30756 37586 393 2202 15934 4643 2206 N N NA
chromosome_1:125835-126254 Cluster_3 chromosome_1 125835 126254 420 5946 24 0.0 - CGAAAACGAAUCUGCUAA 3302 5885 0 59 2 0 0 N N NA
chromosome_1:187614-188042 Cluster_4 chromosome_1 187614 188042 429 942 156 0.024416135881104035 - UAAAUCGGUUUUCUGUUUUCCAACGUGU 279 8 923 3 1 3 4 N N NA
chromosome_1:734332-734761 Cluster_5 chromosome_1 734332 734761 430 809 87 0.9802224969097652 + ACAACAAGAACUUGCGUUUGCUGAACGUU 264 1 794 4 1 5 4 N N NA
chromosome_1:772269-772697 Cluster_6 chromosome_1 772269 772697 429 463 71 0.028077753779697623 - UGUUCAAAUCUGUAAGCAUUAACCGAAGC 240 5 451 0 0 1 6 N N NA
chromosome_1:809191-809619 Cluster_7 chromosome_1 809191 809619 429 1150 216 0.0017391304347826088 - UCAUAUCGUCUACAUUGUGAUGCACCAGU 250 23 1113 0 1 1 12 N N NA
chromosome_1:811151-811576 Cluster_8 chromosome_1 811151 811576 426 1399 392 0.0035739814152966403 - UAGCCGACCAGGAUUGAAGGAUUGAGUCUU 49 19 1352 9 2 1 16 N N NA
chromosome_1:812845-813280 Cluster_9 chromosome_1 812845 813280 436 2670 414 0.0026217228464419477 - UGCUUCUGGUGCACUGUAAAUGACAGCUCC 739
wc -l Results.txt
9259 Results.txt
Columns of interest:
- Column 1 - genomic region of miRNA match
- Column 20 - shortstack miRNA? Y/N
- Column 21 - match to mirbase? NA or mirbase match
awk '{print $1"\t"$20"\t"$21}' "Results.txt" | head | column -t
Locus MIRNA known_miRNAs
chromosome_1:18460-18882 N NA
chromosome_1:70293-70717 N NA
chromosome_1:125835-126254 N NA
chromosome_1:187614-188042 N NA
chromosome_1:734332-734761 N NA
chromosome_1:772269-772697 N NA
chromosome_1:809191-809619 N NA
chromosome_1:811151-811576 N NA
chromosome_1:812845-813280 N NA
miRNAs matching mirbase:
awk '$20 == "Y" && $21 != "NA" {print $1"\t"$20"\t"$21}' "Results.txt" | head | column -t
column: line 3 is too long, output will be truncated
chromosome_6:21902734-21902826 Y nve-miR-9425_MIMAT0035384_Nematostella_vectensis_miR-9425;miR-9425_Nematostella_vectensis_Moran_et_al._2014_NA
chromosome_7:2303817-2303911 Y sca-nve-F-miR-2036_Scolanthus_callimorphus_Praher_et_al._2021_Transcriptome-level;eca-nve-F-miR-2036_Edwardsiella_carnea_Praher_et_al._2021_Transcriptome-level
0-5p;ccr-miR-100_MIMAT0026195_Cyprinus_carpio_miR-100;pmi-miR-100-5p_MIMAT0032156_Patiria_miniata_miR-100-5p;cpi-miR-100-5p_MIMAT0037714_Chrysemys_picta_miR-100-5p;chi-miR-100-5p_MIMAT0035897_Capra_hircus_miR-100-5p;dma-miR-100_MIMAT0049252_Daubentonia_madagascariensis_miR-100;sbo-miR-100_MIMAT0049501_Saimiri_boliviensis_miR-100;ola-miR-100_MIMAT0022614_Oryzias_latipes_miR-100
chromosome_8:4117884-4117974 Y Adi-Mir-2030_5p_Acropora_digitifera_Gajigan_&_Conaco_2017_nve-miR-2030-5p;_nve-miR-2030-5p;_spi-miR-temp-40;ami-nve-F-miR-2030-5p_Acropora_millepora_Praher_et_al._2021_NA;adi-nve-F-miR-2030_Acropora_digitifera__Praher_et_al._2021_NA;spi-mir-temp-40_Stylophora_pistillata_Liew_et_al._2014_Close_match_of_nve-miR-2030;eca-nve-F-miR-2030_Edwardsiella_carnea_Praher_et_al._2021_Transcriptome-level;miR-2030_Nematostella_vectensis_Moran_et_al._2014_NA
chromosome_14:8601339-8601434 Y apa-mir-2037_Exaiptasia_pallida_Baumgarten_et_al._2017_miR-2037;_Nve;_Spis;spi-mir-temp-20_Stylophora_pistillata_Liew_et_al._2014_NA;eca-nve-F-miR-2037_Edwardsiella_carnea_Praher_et_al._2021_Transcriptome-level;spi-nve-F-miR-2037_Stylophora_pistillata_Praher_et_al._2021_NA;sca-nve-F-miR-2037-3p_Scolanthus_callimorphus_Praher_et_al._2021_Transcriptome-level;ami-nve-F-miR-2037-3p_Acropora_millepora_Praher_et_al._2021_NA;mse-nve-F-miR-2037-3p_Metridium_senile_Praher_et_al._2021_Transcriptome-level;avi-miR-temp-2037_Anemonia_viridis_Urbarova_et_al._2018_NA
awk '$20 == "Y" && $21 != "NA" {print $1"\t"$20"\t"$21}' "Results.txt" | wc -l
5
There are 5 miRNAs that had matches to mirbase (all from the cnidarian miRNAs). How many miRNAs were identified in total?
awk '$20 == "Y" {print $1"\t"$20"\t"$21}' "Results.txt" | wc -l
51
Look at fasta
grep "^>" "mir.fasta" | head
>Cluster_30::chromosome_1:2984896-2984988(+)
>Cluster_30.mature::chromosome_1:2984918-2984940(+)
>Cluster_30.star::chromosome_1:2984947-2984968(+)
>Cluster_137::chromosome_1:16932584-16932677(-)
>Cluster_137.mature::chromosome_1:16932635-16932657(-)
>Cluster_137.star::chromosome_1:16932604-16932626(-)
>Cluster_247::chromosome_2:2155517-2155610(+)
>Cluster_247.mature::chromosome_2:2155539-2155561(+)
>Cluster_247.star::chromosome_2:2155568-2155590(+)
>Cluster_449::chromosome_2:20014601-20014694(+)
The fasta starting coordinates need to be fixed due to a big in the code (see issue), which incorrectly calculates the starting coordinates in the fasta output. All other files where start/stop coordinates are displayed are correct. Using Sam White code.
awk '
/^>/ {
# Split the line into main parts based on "::" delimiter
split($0, main_parts, "::")
# Extract the coordinate part and strand information separately
coordinates_strand = main_parts[2]
split(coordinates_strand, coord_parts, "[:-]")
# Determine if the strand information is present and extract it
strand = ""
if (substr(coordinates_strand, length(coordinates_strand)) ~ /[\(\)\-\+]/) {
strand = substr(coordinates_strand, length(coordinates_strand) - 1)
coordinates_strand = substr(coordinates_strand, 1, length(coordinates_strand) - 2)
split(coordinates_strand, coord_parts, "[:-]")
}
# Increment the starting coordinate by 1
new_start = coord_parts[2] + 1
# Reconstruct the description line with the new starting coordinate
new_description = main_parts[1] "::" coord_parts[1] ":" new_start "-" coord_parts[3] strand
# Print the modified description line
print new_description
# Skip to the next line to process the sequence line
next
}
# For sequence lines, print them as-is
{
print
}
' "mir.fasta" \
> "mir_coords_fixed.fasta"
diff "mir.fasta" \
"mir_coords_fixed.fasta" \
| head
< >Cluster_30::chromosome_1:2984896-2984988(+)
---
> >Cluster_30::chromosome_1:2984897-2984988(+)
3c3
< >Cluster_30.mature::chromosome_1:2984918-2984940(+)
---
> >Cluster_30.mature::chromosome_1:2984919-2984940(+)
5c5
< >Cluster_30.star::chromosome_1:2984947-2984968(+)
Success. Select only mature miRNAs from fasta
awk '/^>/ {p = ($0 ~ /mature/)} p' mir_coords_fixed.fasta > mir_coords_fixed_mature.fasta
zgrep -c ">" mir_coords_fixed_mature.fasta
51
I want to double check my 3’UTR identification for Astrangia and rerun miranda with the shortstack miRNAs.
Going to redo the 3’UTR estimation (using e5 deep dive code). I already have all_features.txt, apoc.Chromosome_lenghts.txt, and apoc.Chromosome_names.txt. Generate individual gff for genes
cd /data/putnamlab/jillashey/Astrangia_Genome
grep $'\tgene\t' apoculata_v2.0.gff3 > apoc_gene.gtf
Create 1kb 3’UTR
interactive
module load BEDTools/2.30.0-GCC-11.3.0
bedtools flank -i apoc_gene.gtf -g apoc.Chromosome_lenghts.txt -l 0 -r 1000 -s | awk '{gsub("gene","3prime_UTR",$3); print $0 }' | awk '{if($5-$4 > 3)print $1"\t"$2"\t"$3"\t"$4"\t"$5"\t"$6"\t"$7"\t"$8"\t"$9}' | tr ' ' '\t' > apoc_3UTR_1kb.gtf
Subtract portions of 3’ UTRs that overlap nearby genes
bedtools subtract -a apoc_3UTR_1kb.gtf -b apoc_gene.gtf > apoc_3UTR_1kb_corrected.gtf
This might have more lines because it is subtracting gene overlaps from the 3’UTRs. So if a 3’UTR contains a short gene (200bp), it will subtract that 200bp section and keep the rest of the 3’UTR regardless of where it is. This might not make sense except in my head.
Extract 3’UTR sequences from genome
awk '{print $1 "\t" $4-1 "\t" $5 "\t" $9 "\t" "." "\t" $7}' apoc_3UTR_1kb_corrected.gtf | sed 's/"//g' > apoc_3UTR_1kb_corrected.bed
bedtools getfasta -fi apoculata.assembly.scaffolds_chromosome_level.fasta -bed apoc_3UTR_1kb_corrected.bed -fo apoc_3UTR_1kb.fasta -name
Hooray now we have a fasta of the 3’UTRs with their corresponding gene!
Before running miranda, clean up /data/putnamlab/jillashey/Astrangia2021/smRNA
cd /data/putnamlab/jillashey/Astrangia2021/smRNA
ls -othr
total 1.9G
drwxr-xr-x 4 jillashey 4.0K Oct 9 2023 fastqc
-rw-r--r-- 1 jillashey 80K Jan 4 2024 reads_collapsed.fa
-rw-r--r-- 1 jillashey 92K Jan 4 2024 reads_collapsed_vs_genome.arf
drwxr-xr-x 2 jillashey 4.0K Jan 4 2024 dir_prepare_signature1704403591
-rw-r--r-- 1 jillashey 377 Jan 4 2024 error_04_01_2024_t_16_26_17.log
-rw-r--r-- 1 jillashey 4.5K Jan 4 2024 result_04_01_2024_t_16_26_17.csv
-rw-r--r-- 1 jillashey 45K Jan 4 2024 result_04_01_2024_t_16_26_17.html
drwxr-xr-x 2 jillashey 4.0K Jan 4 2024 pdfs_04_01_2024_t_16_26_17
-rw-r--r-- 1 jillashey 941 Jan 4 2024 result_04_01_2024_t_16_26_17.bed
drwxr-xr-x 2 jillashey 4.0K Jan 4 2024 mirna_results_04_01_2024_t_16_26_17
drwxr-xr-x 2 jillashey 4.0K Jan 4 2024 dir_prepare_signature1704404056
-rw-r--r-- 1 jillashey 377 Jan 4 2024 error_04_01_2024_t_16_34_08.log
-rw-r--r-- 1 jillashey 4.5K Jan 4 2024 result_04_01_2024_t_16_34_08.csv
-rw-r--r-- 1 jillashey 45K Jan 4 2024 result_04_01_2024_t_16_34_08.html
drwxr-xr-x 2 jillashey 4.0K Jan 4 2024 pdfs_04_01_2024_t_16_34_08
-rw-r--r-- 1 jillashey 941 Jan 4 2024 result_04_01_2024_t_16_34_08.bed
drwxr-xr-x 2 jillashey 4.0K Jan 4 2024 mirna_results_04_01_2024_t_16_34_08
-rw-r--r-- 1 jillashey 500M Jan 7 2024 20240107_reads_collapsed.fa
-rw-r--r-- 1 jillashey 147M Jan 7 2024 20240107_reads_collapsed_vs_genome.arf
drwxr-xr-x 7 jillashey 4.0K Jan 7 2024 mirdeep_runs
drwxr-xr-x 2 jillashey 4.0K Jan 7 2024 dir_prepare_signature1704668491
-rw-r--r-- 1 jillashey 29 Jan 7 2024 error_07_01_2024_t_17_59_09.log
-rw-r--r-- 1 jillashey 2.7K Jan 7 2024 report.log
-rw-r--r-- 1 jillashey 449M Jan 16 2024 20240116_reads_collapsed.fa
-rw-r--r-- 1 jillashey 256M Jan 16 2024 20240116_reads_collapsed_vs_genome.arf
drwxr-xr-x 2 jillashey 4.0K Jan 19 2024 mapper_logs
-rw-r--r-- 1 jillashey 359M Jan 19 2024 20240119_reads_collapsed.fa
-rw-r--r-- 1 jillashey 246 Jan 19 2024 bowtie.log
-rw-r--r-- 1 jillashey 181M Jan 19 2024 20240119_reads_collapsed_vs_genome.arf
drwxr-xr-x 3 jillashey 4.0K Jan 24 2024 refs
drwxr-xr-x 22 jillashey 4.0K Jan 30 2024 mirdeep2
drwxr-xr-x 4 jillashey 4.0K Mar 20 2024 data
drwxr-xr-x 2 jillashey 4.0K May 8 2024 miranda
drwxr-xr-x 18 jillashey 4.0K Oct 27 13:52 scripts
drwxr-xr-x 3 jillashey 4.0K Nov 14 08:59 shortstack
mv 2024* mirdeep2
mv reads* mirdeep2
mv result_04_01_2024_t_16_* mirdeep2
mv mapper_logs/ mirdeep2
mv dir_prepare_signature1704* mirdeep2
mv mirna_results_04_01_2024_t_16_* mirdeep2
mv *log mirdeep2
mv pdfs_04_01_2024_t_16_* mirdeep2
mv mirdeep_runs/ mirdeep2
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda
mkdir old
mv * old
Rerun miranda with shortstack mirnas and 3’UTR fasta (with gene names included in fasta headers yay). In the scripts folder, I have a script miranda_strict_all.sh that ran miranda with the mirdeep2 miRNAs. Going to rename this script:
mv miranda_strict_all.sh miranda_strict_all_mirdeep2.sh
Write new script for miranda with shortstack miRNAs. In the scripts folder: nano miranda_strict_all_shortstack.sh
#!/bin/bash -i
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Apoc starting miranda run with all genes and miRNAs with energy cutoff <-20 and strict binding invoked"$(date)
echo "Using updated miRNAs from short stack"
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/shortstack/mir_coords_fixed_mature.fasta /data/putnamlab/jillashey/Astrangia_Genome/apoc_3UTR_1kb.fasta -en -20 -strict -out /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_1kb_apoc_shortstack.tab
conda deactivate
echo "miranda run finished!" $(date)
echo "counting number of interactions attempted" $(date)
zgrep -c "Performing Scan" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_1kb_apoc_shortstack.tab
echo "Parsing output" $(date)
grep -A 1 "Scores for this hit:" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_1kb_apoc_shortstack.tab | sort | grep '>' > /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_1kb_apoc_shortstack_parsed.txt
echo "counting number of putative interactions predicted" $(date)
wc -l /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_1kb_apoc_shortstack_parsed.txt
echo "Apoc miranda script complete" $(date)
Submitted batch job 348808. Ran in ~20 mins. Look at output:
counting number of interactions attempted Thu Nov 14 14:43:19 EST 2024
2547144
Parsing output Thu Nov 14 14:43:28 EST 2024
counting number of putative interactions predicted Thu Nov 14 14:43:28 EST 2024
5187 /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_1kb_apoc_shortstack_parsed.txt
Look at txt file
head miranda_strict_all_1kb_apoc_shortstack_parsed.txt
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_10.126;Name=EVM%20prediction%20chromosome_10.126::chromosome_10:1142345-1143345 170.00 -21.16 2 20 717 739 19 84.21% 84.21%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_11.3016;Name=EVM%20prediction%20chromosome_11.3016::chromosome_11:31902611-31903611 166.00 -21.91 2 21 842 865 21 76.19% 85.71%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_11.359;Name=EVM%20prediction%20chromosome_11.359::chromosome_11:3751965-3752965 164.00 -20.30 2 18 439 461 17 82.35% 88.24%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_12.1475;Name=EVM%20prediction%20chromosome_12.1475::chromosome_12:15544468-15545468 155.00 -21.98 2 17 149 174 19 73.68% 78.95%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_12.1806;Name=EVM%20prediction%20chromosome_12.1806::chromosome_12:19406302-19407302 163.00 -20.09 2 17 977 999 16 87.50% 87.50%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_12.2475;Name=EVM%20prediction%20chromosome_12.2475::chromosome_12:26095701-26096701 173.00 -20.76 2 21 336 355 19 84.21% 89.47%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_12.2814;Name=EVM%20prediction%20chromosome_12.2814::chromosome_12:28905929-28906929 163.00 -20.09 2 17 349 371 16 87.50% 87.50%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_13.1892;Name=EVM%20prediction%20chromosome_13.1892::chromosome_13:19643825-19644825 177.00 -20.79 2 18 860 881 16 93.75% 93.75%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_13.2239;Name=EVM%20prediction%20chromosome_13.2239::chromosome_13:23509872-23510872 173.00 -21.02 2 21 577 596 19 84.21% 89.47%
>Cluster_1155.mature::chromosome_5:8149619-8149640(-) ID=evm.TU.chromosome_13.330;Name=EVM%20prediction%20chromosome_13.330::chromosome_13:2959926-2960926 172.00 -21.83 2 17 218 239 15 93.33% 93.33%
How many unique miRNAs had predicted interactions?
cut -f1 miranda_strict_all_1kb_apoc_shortstack_parsed.txt | sort | uniq | wc -l
51
How many unique 3’UTRs had interactions with miRNAs?
cut -f2 miranda_strict_all_1kb_apoc_shortstack_parsed.txt | sort | uniq | wc -l
4515
Copy miranda_strict_all_1kb_apoc_shortstack_parsed.txt onto local computer.
20241118
Rerun mirdeep2???? Okay. I feel like I have a little more clarity with the mirdeep2 analysis now that I’ve also run it for my Mcap data. First, I need to map the samples to the genome with the mapper.pl script. Genome must first be indexed by bowtie-build (NOT bowtie2). Because I have multiple samples, I need to make a config file that contains fq file locations and a unique 3 letter code (see mirdeep2 documentation).
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
nano config.txt
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1065_R1_001.fastq.gz_1.fastq s01
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1147_R1_001.fastq.gz_1.fastq s02
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1412_R1_001.fastq.gz_1.fastq s03
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1560_R1_001.fastq.gz_1.fastq s04
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1567_R1_001.fastq.gz_1.fastq s05
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1617_R1_001.fastq.gz_1.fastq s06
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-1722_R1_001.fastq.gz_1.fastq s07
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2000_R1_001.fastq.gz_1.fastq s08
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2007_R1_001.fastq.gz_1.fastq s09
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2302_R1_001.fastq.gz_1.fastq s10
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2360_R1_001.fastq.gz_1.fastq s11
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2398_R1_001.fastq.gz_1.fastq s12
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2404_R1_001.fastq.gz_1.fastq s13
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2412_R1_001.fastq.gz_1.fastq s14
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2512_R1_001.fastq.gz_1.fastq s15
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2523_R1_001.fastq.gz_1.fastq s16
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2563_R1_001.fastq.gz_1.fastq s17
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2729_R1_001.fastq.gz_1.fastq s18
/data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar/AST-2755_R1_001.fastq.gz_1.fastq s19
In the config.txt file, I have the path and file name, as well as the unique 3 letter code (s followed by sample number).
In the scripts folder: nano mapper_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load GCCcore/11.3.0 #I needed to add this to resolve conflicts between loaded GCCcore/9.3.0 and GCCcore/11.3.0
module load Bowtie/1.3.1-GCC-11.3.0
echo "Index Apoc genome" $(date)
# Index the reference genome for Apoc
bowtie-build /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/Apoc_ref.btindex
echo "Referece genome indexed!" $(date)
echo "Unload unneeded packages and run mapper script for trimmed stringent reads" $(date)
module unload module load GCCcore/11.3.0
module unload Bowtie/1.3.1-GCC-11.3.0
conda activate /data/putnamlab/mirdeep2
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
mapper.pl config.txt -e -d -h -j -l 18 -m -p /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/Apoc_ref.btindex -s apoc_mapped_reads.fa -t apoc_mapped_reads_vs_genome.arf
echo "Mapping complete for trimmed reads" $(date)
conda deactivate
Submitted batch job 349330. Ran in about an hour.
20241119
Look at the output
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
zgrep -c ">" apoc_mapped_reads.fa
88460783
wc -l apoc mapped_reads_vs_genome.arf
17920088 apoc_mapped_reads_vs_genome.arf
less bowtie.log
# reads processed: 4816214
# reads with at least one reported alignment: 326607 (6.78%)
# reads that failed to align: 4413294 (91.63%)
# reads with alignments suppressed due to -m: 76313 (1.58%)
Reported 624511 alignments to 1 output stream(s)
I can now run mirdeep2 to predict my miRNAs. Move the mapper output from the data folder to the output folder.
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2
mkdir 20241119
cd /data/putnamlab/jillashey/Astrangia2021/smRNA/data/trim/flexbar
mv mapper_logs/ /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/20241119/
mv *mapped* /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/20241119/
mv bowtie.log /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/20241119/
The output files from the mapper.pl portion of mirdeep2 will be used as input for the mirdeep2.pl portion of the pipeline. In the scripts folder: nano mirna_predict_mirdeep2.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/mirdeep2
echo "Starting mirdeep2" $(date)
miRDeep2.pl /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/20241119/apoc_mapped_reads.fa /data/putnamlab/jillashey/Astrangia_Genome/apoculata.assembly.scaffolds_chromosome_level.fasta /data/putnamlab/jillashey/Astrangia2021/smRNA/mirdeep2/20241119/apoc_mapped_reads_vs_genome.arf /data/putnamlab/jillashey/Astrangia2021/smRNA/refs/mature_mirbase_cnidarian_T.fa none none -P -v -g -1 2>report.log
echo "mirdeep2 complete" $(date)
conda deactivate
Submitted batch job 349437. Ran in about 2 days.
20241121
Downloaded output to computer and identified putative novel and known miRNAs. There were ~240 putative miRNAs total. I will need to look at pdfs to determine MFE and total number of binding bps in hairpin structure.
Went with shortstack.
Map miRNAs to CDS.
In the scripts folder: nano miranda_strict_mrna_shortstack.sh
#!/bin/bash -i
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Astrangia2021/smRNA/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Apoc starting miranda run with all genes and miRNAs with energy cutoff <-20 and strict binding invoked"$(date)
echo "Using updated miRNAs from short stack"
echo "Using mRNA sequences as miRNA targets"
#module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/miranda
miranda /data/putnamlab/jillashey/Astrangia2021/smRNA/shortstack/mir_coords_fixed_mature.fasta /data/putnamlab/jillashey/Astrangia_Genome/apoculata_mrna_v2.0.fasta -en -20 -strict -out /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_mrna_apoc_shortstack.tab
conda deactivate
echo "miranda run finished!" $(date)
echo "counting number of interactions attempted" $(date)
zgrep -c "Performing Scan" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_mrna_apoc_shortstack.tab
echo "Parsing output" $(date)
grep -A 1 "Scores for this hit:" /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_mrna_apoc_shortstack.tab | sort | grep '>' > /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_mrna_apoc_shortstack_parsed.txt
echo "counting number of putative interactions predicted" $(date)
wc -l /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_mrna_apoc_shortstack_parsed.txt
echo "Apoc miranda script complete" $(date)
Submitted batch job 365009. Results:
counting number of interactions attempted Mon May 5 14:52:16 EDT 2025
2457384
Parsing output Mon May 5 14:52:23 EDT 2025
counting number of putative interactions predicted Mon May 5 14:52:24 EDT 2025
12731 /data/putnamlab/jillashey/Astrangia2021/smRNA/miranda/miranda_strict_all_mrna_apoc_shortstack_parsed.txt