
Apulchra genome assembly
Apulchra genome assembly
Sperm and tissue from adult Acropora pulchra colonies were collected from Moorea, French Polynesia and sequencing with PacBio (long reads) and Illumina (short reads). This post will detail the genome assembly notes. The github for this project is here.
I’m going to write notes and code chronologically so that I can keep track of what I’m doing each day. When assembly is complete, I will compile the workflow in a separate post.
20240206
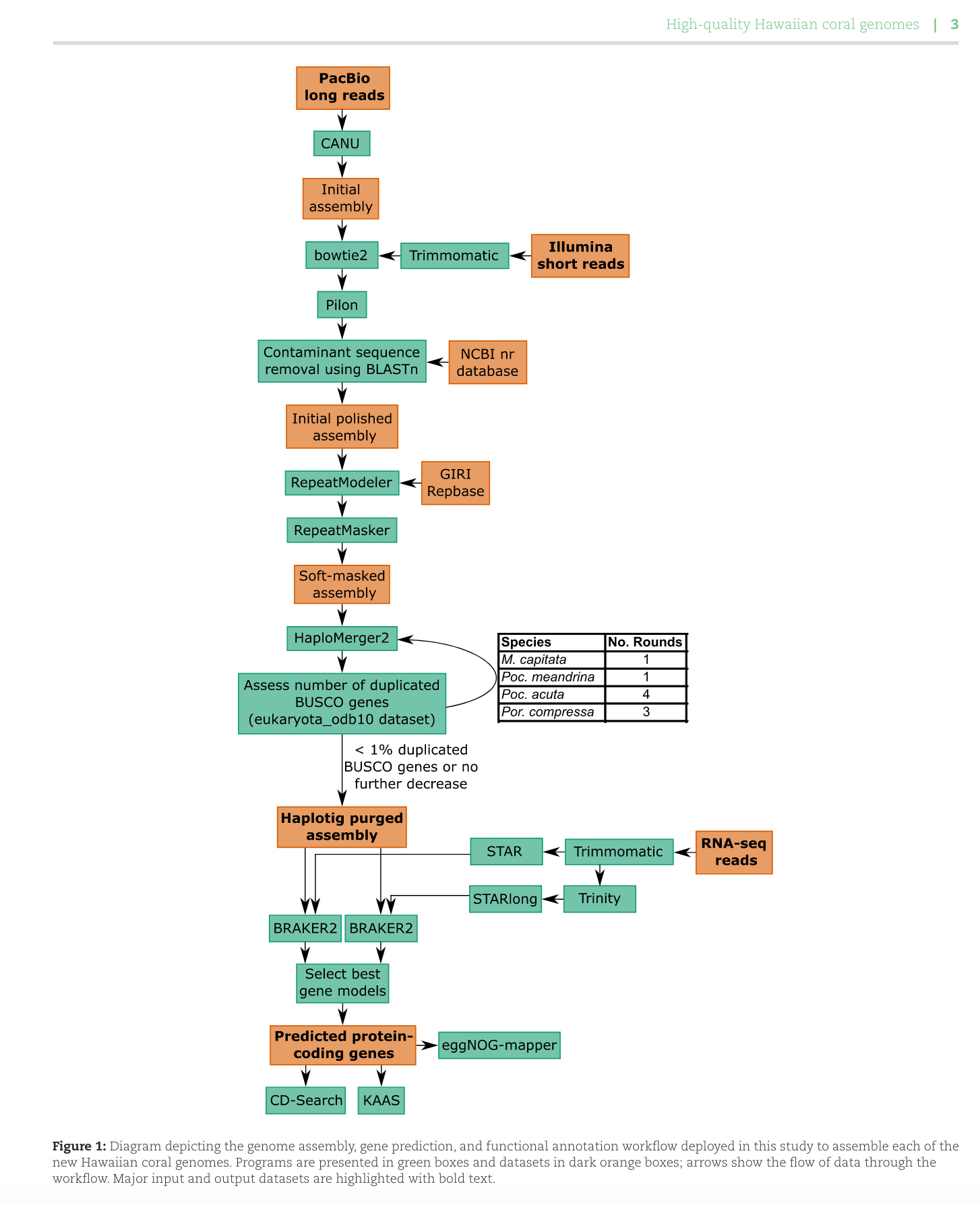
Met w/ Ross and Hollie today re Apulchra genome assembly. We decided to move forward with the workflow from Stephens et al. 2022 which assembled genomes for 4 Hawaiian corals.
PacBio long reads were received in late Jan/early Feb 2024. According to reps from Genohub, the PacBio raw output looks good.
We decided to move forward with Canu to assembly the genome. Canu is specialized to assemble PacBio sequences, operating in three phases: correction, trimming and assembly. According to the Canu website, “The correction phase will improve the accuracy of bases in reads. The trimming phase will trim reads to the portion that appears to be high-quality sequence, removing suspicious regions such as remaining SMRTbell adapter. The assembly phase will order the reads into contigs, generate consensus sequences and create graphs of alternate paths.”
The PacBio files that will be used for assembly are located here on Andromeda: /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead. The files in the folder that we will use are:
m84100_240128_024355_s2.hifi_reads.bc1029.bam
m84100_240128_024355_s2.hifi_reads.bc1029.bam.pbi
The bam file contains all of the read information in computer language and the pbi file is an index file of the bam. Both are needed in the same folder to run Canu.
For Canu, input files must be fasta or fastq format. I’m going to use bam2fastqfrom the PacBio github. This module is not on Andromeda so I will need to install it via conda.
The PacBio sequencing for the Apul genome were done with HiFi sequencing that are produced with circular consensus sequencing on PacoBio long read systems. Here’s how HiFi reads are generated from the PacBio website:
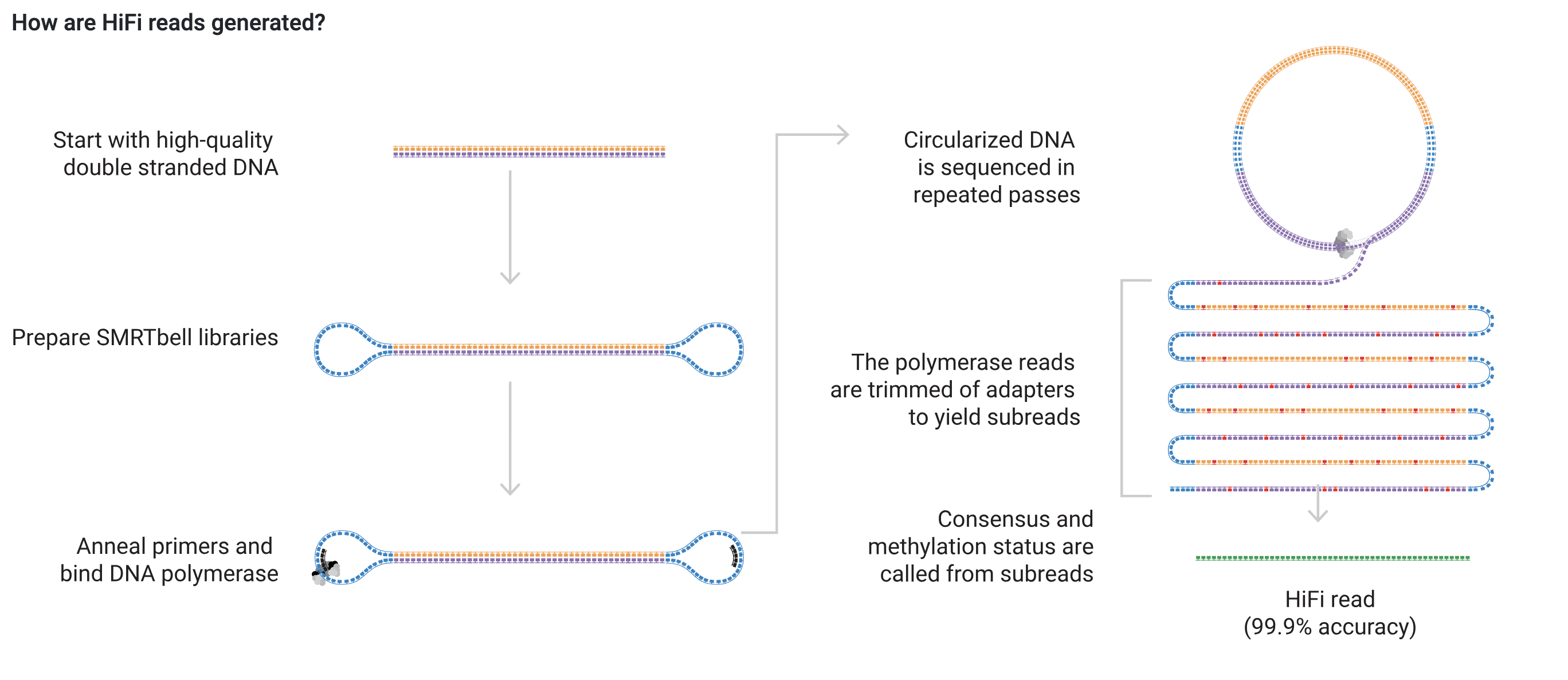
Since Hifi sequencing was used, a specific HiCanu flag (-pacbio-hifi) must be used. Additionally, in the Canu tutorial, it says that if this flag is used, the program is assuming that the reads are trimmed and corrected already. However, our reads are not. I’m going to try to run the first pass at Canu with the -raw flag.
Before Canu, I will run bam2fastq. This is a part of the PacBio BAM toolkit package pbtk. I need to create the conda environment and install the package. Load miniconda module: module load Miniconda3/4.9.2. Create a conda environment.
conda create --prefix /data/putnamlab/conda/pbtk
Install the package. Once the package is installed on the server, anyone in the Putnam lab group can use it.
conda activate /data/putnamlab/conda/pbtk
conda install -c bioconda pbtk
In my own directory, make a new directory for genome assembly
cd /data/putnamlab/jillashey
mkdir Apul_Genome
cd Apul_Genome
mkdir assembly structural functional
cd assembly
mkdir scripts data output
Run code to make the PacBio bam file to a fastq file. In the scripts folder: nano bam2fastq.sh
#!/bin/bash -i
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/pbtk
echo "Convert PacBio bam file to fastq file" $(date)
bam2fastq -o /data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.fastq /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029.bam
echo "Bam to fastq complete!" $(date)
conda deactivate
Submitted batch job 294235
20240208
Job pended for about a day, then ran in 1.5 hours. I got this error message: bash: cannot set terminal process group (-1): Function not implemented bash: no job control in this shell, but not sure why. A fastq.gz file was produced! The file is pretty large (35G).
less m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq.gz
@m84100_240128_024355_s2/261887593/ccs
TATAAGTTTTACAGCTGCCTTTTGCTCAGCAAAGAAAGCAGCATTGTTATTGAACAGAAAAAGCCTTTTGGTGATATAAAGGTTTCTAAGGGACCAAAGTTTGATCTAGTATGCTAAGTGTGGTGGGTTAAAACTTTGTTTCACCTTTTTCCCGTGATGTTACAAAATTGGTGCAAATATTCATGACGTCGTACTCAAGTCTGACACTAAGAGCATGCAATTACTTAAACAAACAAGCCATACACCAATAAACTGAAGCTCTGTCAACTAGAAAACCTTTGAGTATTTTCATTGTAAAGTACAAGTGGTATATTGTCACTTGCTTTTACAACTTGAAGAACTCACAGTTAAGTTACTAGATTCACCATAGTGCTTGGCAATGAAGAAGCCAAATCACATAAAGTCGGAGCATGTGGTGTTTAGACCTAATCAAACAAGAACACAATATTTAGTACCTGCATCCTGTCTAAGGAGGAAATTTTAAGCTGCTTTCTTTTAAATTTTTTTTATTAGCATTTCAATGGTTGAGGTCGATTATAGGTGCTAGGCTTTAATTCCGTACTATGAAAGAAGAAAGGTCGTTGTTATTAACCATGTCAAACAGAGAAACACATGGTAAAAAATTGACTTCCTTTTCCTCTCGTTGCCACTTAAGCTTAATGATGGTGTTTGACCTGAAAGATGTTACAATTGTTTTAGATGAAAAGACTGTTCTGCGTAAAACAGTGAAGCCTCCCAACTTATTTTGTTATGTGGATTTTGTTGTCTTGTTAGTAACATGTATTGGACTATCTTTTGTGAGTACATAGCTTTTTTTCCATCAACTGACTATATACGTGGTGTAATTTGAGATCATGCCTCCAAGTGTTAGTCTTTTGTTTGGGGCTAACTCGTAAAAGACAAAGGGAGGGGGGTTGTCTAATTCCTAAGCAAAGCATTAAGTTTAACACAGGAAATTGTTTGCGTTGATATTGCTATCCTTTCAGCCCCAAACAAAAAATTTAATGGTTATTTTATTTTACATCTATTGTAAATATATTTTAACATTAATTTTTATTATTGCACTGTAAATACTTGTACTAATGTTCTGTTTGAATTAATTTTGATTCATTCCTTGTGCTTACAACAACAGGGATACAAAACCGATATGTATAATAATACTATTAGAGATGCTTATTTGCATTTTTAGCCCATACCATGAGTTTTAATAACGCCAGGCCATTGGAGATTTTATGGAGTGAGGATTCATTGTACAAACATGGTTGATTTAATATTAAAGTTGTATCCAAATAATTAATATCTGCTGTGATCAGTGAAAGATTGACCTTTCAGTTGTTTGGTTGCACCTTCATCTTATTGGAAACAACTGAATGGAGCATCTTTCCAGTTTAAAAATGTACCACTGCCCACTTTCATGAAGTTATGCCACATATTAATAATGACTATTAATTGTTGAAAACCCTTCTTCCAAAATGTTTCCATTTATTTGTAATAGCATATGTGGTCCATCAGAACAATAATTTAAATCATTACTATTAATAATTTTCCAATAACTGACTTTCAAACCTAGCCAACAAGCATAAGTCAGTAAGCCACAGAGTCCAGAGATACACTTACACTTTACTTTTCACTTCTGAAACATTTTATAATCTCAGTATGAGCATAGAACTTTTCAGTTGGGCAGCATGGAATAGAACCTTTGGACCCCTCTGTGAATATCAAAAATAGGCAACCACTTCCAGCATACACTCTAGCCTCCTTCATAAAGCAAGCCTTAGTGTTTTAGCTTCTACTAGTTAGATTCATTTTAAAAGAAGTTCAGTATACTTAATCTTATAGAAGCTGATTGTGATATAATTGCATAGGTGGATCTCAGAAAAGTGAGATGTAGCTGTCAAATTAAAAGAAGTCCTTTCCAAGCGTAGCTTCTGATAAACAATGCATTTTAGTTAACATTGGATTATGGTTTCAAAGGACTTGTAAAGCTAAATTCAAGTTTTTATGACAACTTGAAAGCCTTTGCCACAGTCTCCGCTGATTTAAGACTTCCATCAAAGTTAGAGTGGTGTGAATGCATCTCCACATGCAATTAATAAAGGTGAGGCAGACAACACAAAACACCCTGGTGCACCATCAACTCCACGGATCACTTGACTGTAACGCCATCTTATACAGCGACTGCCAATTGGAACTGGAAGATCAGGAATGATCTCTTTCACATGGGAAATGAGCATGGTCTTGATAATGCTTTCATCAGTATCCCAATGTTGAAGACAGAAGGACTTTGTTGAATGTACTAAAAGAGAGGGACCAACATCCACTGGATCTGGAACAGAATGCCAAAGCATATGCAAAATGTTATCATGTATAATAAAGAAAAAACCAAACTCATGAAGTTCAACGGTCACAGGCTTTGGTAAAAGACTGTACATGGTAAAGTACTCAGGGTGAAAGTTTGTATCCAAAAAACGAAAAGCTAACCTGTACCACATCTCTTCCGAGAGTCCACTGACACAAATGCAATATTAGGATTGCCAGTCGCATATTTACACGTCCATGGCACATTGATAACTGCTTCATGATCAAAAAAGTATCCTACTGCAAACCGTGATGAATATTCCACATTTTGTAAGGATTTGATTTCATTTTGTAAGAATGCTTGAATTGAACCTTGTAGCTGAAGAAGTTGTGGTACTGGAATAGTTACTATGACTGA
See how many @m84100 are in the file. I’m not sure what these stand for, maybe contigs?
zgrep -c "@m84100" m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq.gz
5898386
More than 5 million, so many not contigs? I guess it represents the number of HiFi reads generated. Now time to run Canu! Canu is already installed on the server, which is nice.
In the scripts folder: nano canu.sh
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load canu/2.2-GCCcore-11.2.0
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Unzip paco-bio fastq file" $(date)
gunzip m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq.gz
echo "Unzip complete, starting assembly" $(date)
canu -p apul -d /data/putnamlab/jillashey/Apul_Genome/assembly/data genomeSize=475m -raw -pacbio-hifi m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq
echo "Canu assembly complete" $(date)
I’m not sure if the raw and -pacbio-hifi will be compatible, as the Canu tutorial says that the -pacbio-hifi assumes that the input is trimmed and corrected (still not sure what this means). Submitted batch job 294325
20240212
The canu script appears to have ran. The script canu.sh itself ran in ~40 mins, but it spawned 10000s of other jobs on the server (for parallel processing, I’m guessing). Since I didn’t start those jobs, I didn’t get emails when they finished, so I just had to check the server every few hours. It took about a day for the rest of the jobs to finish. First, I’m looking at the slurm-294325.error output file. There’s a lot in this file, but I will break it down.
It first provides details on the slurm support and associated memory, as well as the number of threads that each portion of canu will need to run.
-- Slurm support detected. Resources available:
-- 25 hosts with 36 cores and 124 GB memory.
-- 3 hosts with 24 cores and 124 GB memory.
-- 1 host with 36 cores and 123 GB memory.
-- 1 host with 48 cores and 753 GB memory.
-- 8 hosts with 36 cores and 61 GB memory.
-- 1 host with 48 cores and 250 GB memory.
-- 2 hosts with 36 cores and 250 GB memory.
-- 2 hosts with 48 cores and 375 GB memory.
-- 4 hosts with 36 cores and 502 GB memory.
--
-- (tag)Threads
-- (tag)Memory |
-- (tag) | | algorithm
-- ------- ---------- -------- -----------------------------
-- Grid: meryl 12.000 GB 6 CPUs (k-mer counting)
-- Grid: hap 12.000 GB 12 CPUs (read-to-haplotype assignment)
-- Grid: cormhap 13.000 GB 12 CPUs (overlap detection with mhap)
-- Grid: obtovl 8.000 GB 6 CPUs (overlap detection)
-- Grid: utgovl 8.000 GB 6 CPUs (overlap detection)
-- Grid: cor -.--- GB 4 CPUs (read correction)
-- Grid: ovb 4.000 GB 1 CPU (overlap store bucketizer)
-- Grid: ovs 16.000 GB 1 CPU (overlap store sorting)
-- Grid: red 10.000 GB 6 CPUs (read error detection)
-- Grid: oea 8.000 GB 1 CPU (overlap error adjustment)
-- Grid: bat 64.000 GB 8 CPUs (contig construction with bogart)
-- Grid: cns -.--- GB 8 CPUs (consensus)
--
-- Found trimmed raw PacBio HiFi reads in the input files.
The file also says that it skipped the correction and trimming steps. This indicates that adding the -raw flag didn’t work.
-- Stages to run:
-- assemble HiFi reads.
--
--
-- Correction skipped; not enabled.
--
-- Trimming skipped; not enabled.
Two histograms are then presented. The first is a histogram of correct reads:
-- In sequence store './apul.seqStore':
-- Found 5897694 reads.
-- Found 79182880880 bases (166.7 times coverage).
-- Histogram of corrected reads:
--
-- G=79182880880 sum of || length num
-- NG length index lengths || range seqs
-- ----- ------------ --------- ------------ || ------------------- -------
-- 00010 26133 264284 7918288640 || 1150-2287 8297|--
-- 00020 22541 592551 15836596549 || 2288-3425 76768|-------------
-- 00030 20184 964605 23754880672 || 3426-4563 316378|---------------------------------------------------
-- 00040 18285 1377072 31673163019 || 4564-5701 397324|---------------------------------------------------------------
-- 00050 16551 1832151 39591448183 || 5702-6839 374507|------------------------------------------------------------
-- 00060 14786 2337786 47509730799 || 6840-7977 351843|--------------------------------------------------------
-- 00070 12854 2910882 55428020435 || 7978-9115 339864|------------------------------------------------------
-- 00080 10612 3585762 63346313974 || 9116-10253 340204|------------------------------------------------------
-- 00090 7724 4449769 71264594524 || 10254-11391 341160|-------------------------------------------------------
-- 00100 1150 5897693 79182880880 || 11392-12529 343170|-------------------------------------------------------
-- 001.000x 5897694 79182880880 || 12530-13667 340043|------------------------------------------------------
-- || 13668-14805 336214|------------------------------------------------------
-- || 14806-15943 328927|-----------------------------------------------------
-- || 15944-17081 316290|---------------------------------------------------
-- || 17082-18219 293393|-----------------------------------------------
-- || 18220-19357 261212|------------------------------------------
-- || 19358-20495 225351|------------------------------------
-- || 20496-21633 188701|------------------------------
-- || 21634-22771 154123|-------------------------
-- || 22772-23909 124842|--------------------
-- || 23910-25047 99659|----------------
-- || 25048-26185 78280|-------------
-- || 26186-27323 61530|----------
-- || 27324-28461 47760|--------
-- || 28462-29599 37668|------
-- || 29600-30737 29339|-----
-- || 30738-31875 22548|----
-- || 31876-33013 17292|---
-- || 33014-34151 13055|---
-- || 34152-35289 9797|--
-- || 35290-36427 7251|--
-- || 36428-37565 5005|-
-- || 37566-38703 3560|-
-- || 38704-39841 2474|-
-- || 39842-40979 1553|-
-- || 40980-42117 1006|-
-- || 42118-43255 604|-
-- || 43256-44393 334|-
-- || 44394-45531 184|-
-- || 45532-46669 102|-
-- || 46670-47807 45|-
-- || 47808-48945 18|-
-- || 48946-50083 11|-
-- || 50084-51221 4|-
-- || 51222-52359 1|-
-- || 52360-53497 2|-
-- || 53498-54635 0|
-- || 54636-55773 0|
-- || 55774-56911 0|
-- || 56912-58049 1|-
There’s also a histogram of corrected-trimmed reads, but it is the exact same as the histogram above. The histogram represents the length ranges for each sequence and the number of sequences that have that length range. For example, if we look at the top row, there are 8297 sequences that range in length from 1150-2287 bp.
It looks like canu did run jobs by itself:
-- For 5897694 reads with 79182880880 bases, limit to 791 batches.
-- Will count kmers using 16 jobs, each using 13 GB and 6 threads.
--
-- Finished stage 'merylConfigure', reset canuIteration.
--
-- Running jobs. First attempt out of 2.
--
-- 'meryl-count.jobSubmit-01.sh' -> job 294326 tasks 1-16.
--
----------------------------------------
-- Starting command on Thu Feb 8 14:32:12 2024 with 8923.722 GB free disk space
cd /glfs/brick01/gv0/putnamlab/jillashey/Apul_Genome/assembly/data
sbatch \
--depend=afterany:294326 \
--cpus-per-task=1 \
--mem-per-cpu=4g \
-D `pwd` \
-J 'canu_apul' \
-o canu-scripts/canu.01.out canu-scripts/canu.01.sh
-- Finished on Thu Feb 8 14:32:13 2024 (one second) with 8923.722 GB free disk space
Let’s look at the output files that canu produced in /data/putnamlab/jillashey/Apul_Genome/assembly/data. I’ll be using the Canu tutorial output information to understand outputs.
-rwxr-xr-x. 1 jillashey 1.1K Feb 8 14:13 apul.seqStore.sh
-rw-r--r--. 1 jillashey 951 Feb 8 14:31 apul.seqStore.err
drwxr-xr-x. 3 jillashey 4.0K Feb 8 14:32 apul.seqStore
drwxr-xr-x. 9 jillashey 4.0K Feb 8 23:57 unitigging
drwxr-xr-x. 2 jillashey 4.0K Feb 9 01:36 canu-scripts
lrwxrwxrwx. 1 jillashey 24 Feb 9 01:36 canu.out -> canu-scripts/canu.09.out
drwxr-xr-x. 2 jillashey 4.0K Feb 9 01:36 canu-logs
-rw-r--r--. 1 jillashey 23K Feb 9 01:47 apul.report
-rw-r--r--. 1 jillashey 7.0M Feb 9 01:51 apul.contigs.layout.tigInfo
-rw-r--r--. 1 jillashey 155M Feb 9 01:51 apul.contigs.layout.readToTig
-rw-r--r--. 1 jillashey 2.8G Feb 9 01:57 apul.unassembled.fasta
-rw-r--r--. 1 jillashey 943M Feb 9 02:01 apul.contigs.fasta
The apul.report file will provide information about the analysis during assembly, including histogram of read lengths, the histogram or k-mers in the raw and corrected reads, the summary of corrected data, summary of overlaps, and the summary of contig lengths. The histogram of read lengths is the same as in the error file above. There is also a histogram (?) of the mer information:
-- 22-mers Fraction
-- Occurrences NumMers Unique Total
-- 1- 1 0 0.0000 0.0000
-- 2- 2 4316301 **** 0.0150 0.0002
-- 3- 4 855268 0.0172 0.0002
-- 5- 7 183637 0.0183 0.0002
-- 8- 11 62578 0.0187 0.0002
-- 12- 16 29616 0.0188 0.0002
-- 17- 22 22278 0.0189 0.0003
-- 23- 29 20222 0.0190 0.0003
-- 30- 37 28236 0.0190 0.0003
-- 38- 46 73803 0.0192 0.0003
-- 47- 56 556391 0.0194 0.0003
-- 57- 67 6166302 ****** 0.0218 0.0010
-- 68- 79 35800144 *************************************** 0.0477 0.0098
-- 80- 92 63190280 ********************************************************************** 0.1835 0.0633
-- 93- 106 26607016 ***************************** 0.3988 0.1607
-- 107- 121 2495822 ** 0.4799 0.2024
-- 122- 137 2376701 ** 0.4871 0.2066
-- 138- 154 16117515 ***************** 0.4965 0.2131
-- 155- 172 47510630 **************************************************** 0.5575 0.2605
-- 173- 191 41907409 ********************************************** 0.7262 0.4060
-- 192- 211 9190117 ********** 0.8650 0.5375
-- 212- 232 1806623 ** 0.8934 0.5669
-- 233- 254 4028020 **** 0.8997 0.5743
-- 255- 277 3875382 **** 0.9140 0.5926
-- 278- 301 1456651 * 0.9271 0.6107
-- 302- 326 1905710 ** 0.9319 0.6181
-- 327- 352 3003944 *** 0.9388 0.6293
-- 353- 379 1723658 * 0.9491 0.6477
-- 380- 407 968475 * 0.9549 0.6587
-- 408- 436 1249856 * 0.9583 0.6657
-- 437- 466 890757 0.9626 0.6752
-- 467- 497 768714 0.9656 0.6824
-- 498- 529 937920 * 0.9683 0.6892
-- 530- 562 618907 0.9716 0.6978
-- 563- 596 582755 0.9737 0.7039
-- 597- 631 535408 0.9757 0.7100
-- 632- 667 441122 0.9775 0.7159
-- 668- 704 459953 0.9791 0.7211
-- 705- 742 367266 0.9807 0.7268
-- 743- 781 354455 0.9819 0.7316
-- 782- 821 304354 0.9832 0.7365
There are 22-mers. A k-mer are substrings of length k contained in a biological sequence. For example, the term k-mer refers to all of a sequence’s subsequences of length k such that the sequence AGAT would have four monomers (A, G, A, and T), three 2-mers (AG, GA, AT), two 3-mers (AGA and GAT) and one 4-mer (AGAT). So if we have 22-mers, we have subsequences of 22 nt? The Canu documentation says that k-mer histograms with more than 1 peak likely indicate a heterozygous genome. I’m not sure if the stars represent peaks or counts but if this is a histogram of k-mer information, it has two peaks, indicating a heterozygous genome.
The Canu documentation states that corrected read reports should be given with information about number of reads, coverage, N50, etc. My log file does not have this information, likely because the trimming and correcting steps were not performed. Instead, I have this information:
-- category reads % read length feature size or coverage analysis
-- ---------------- ------- ------- ---------------------- ------------------------ --------------------
-- middle-missing 4114 0.07 10652.45 +- 6328.73 1185.95 +- 2112.86 (bad trimming)
-- middle-hump 4148 0.07 11485.92 +- 4101.92 4734.88 +- 3702.70 (bad trimming)
-- no-5-prime 8533 0.14 9311.86 +- 5288.80 2087.03 +- 3315.79 (bad trimming)
-- no-3-prime 10888 0.18 7663.24 +- 5159.57 1677.51 +- 3090.22 (bad trimming)
--
-- low-coverage 48831 0.83 6823.01 +- 3725.15 16.35 +- 15.52 (easy to assemble, potential for lower quality consensus)
-- unique 5419159 91.89 9403.44 +- 4761.57 110.87 +- 40.33 (easy to assemble, perfect, yay)
-- repeat-cont 93801 1.59 7730.76 +- 4345.55 1001.60 +- 665.30 (potential for consensus errors, no impact on assembly)
-- repeat-dove 380 0.01 23076.97 +- 3235.06 878.08 +- 510.47 (hard to assemble, likely won't assemble correctly or eve
n at all)
--
-- span-repeat 64724 1.10 11397.59 +- 5058.57 2335.58 +- 2836.91 (read spans a large repeat, usually easy to assemble)
-- uniq-repeat-cont 182925 3.10 9764.80 +- 4218.80 (should be uniquely placed, low potential for consensus e
rrors, no impact on assembly)
-- uniq-repeat-dove 14288 0.24 17978.89 +- 4847.85 (will end contigs, potential to misassemble)
-- uniq-anchor 19659 0.33 11312.36 +- 4510.86 5442.58 +- 3930.94 (repeat read, with unique section, probable bad read)
I’m not sure why I’m getting all of this information or what it means. There is a high % of unique reads in the data which is good. In the file, there is also information about edges (not sure what this means), as well as error rates. May discuss further with Hollie.
The Canu output documentation says that I’m supposed to get a file with corrected and trimmed reads but I don’t have those. I do have apul.unassembled.fasta and apul.contigs.fasta.
head apul.unassembled.fasta
>tig00000838 len=19665 reads=4 class=unassm suggestRepeat=no suggestBubble=no suggestCircular=no trim=0-19665
TAAAAACATTGATTCTTGTTTCAATATGAGACTTGTTTCGGAAGATGTTCGCGCAGGTTACATTTCATAATCCACAAGAAATGCGACATCGCCAACCTTA
CTTTAGTGTTTGCATTTAAGCAAAACAATGATAAAGAAACAAATCTCACATCTCGCAAAAGTATGCATTCTATGAAGAACAATGAAAATTAATGAAAGTG
AATCTTACACCTCCTATTCAAGACGCCGCATTAATTCAACTTGTTGATTTCTCCTAAAACGCTTTCTTTTAGAAGGCTTTCTTAGTCTTTCAATTGTAAA
GAATACATAAAGGACTCATGACCACTTATGTTCTTAAGTGTTACTGCTGCTTTAAAACACGTTACAAACCACATGTGAATATAGTTGCGGCACAGAAGGG
AAAATCGCTGAAATGCTGTCCAAATATACACAATATCATTAAGTAAAGTACGATCGTCCGGGTGAGTGTAGTCCTGAGAAGGACTGTTTGAGATGACATT
GACTGACGTTTCGACAACCTGAGCGGAAGTCATCTTCAGAGTCATCTTCACTTGACTCTGAAGATGACTTCCGCTCAGGTTGTCGAAACGTCAGTCAATG
TCATCTCAAACAGTCCTTCTCAGGACTACACTCACCCGGACGATCGTACTTTACTTAATGATATGACTCCTGGGTTCAAACCATTTACAAATATACACAA
GTTTGAAAGATCATATCGCCTGCCAGTTTTACAACTCGTCTTAGACACAATGGAATACAAAACCCTACCGAACGAATACCTTTGATTGAGATTTATGAAT
GTGAAACAGCGACTTCGAGAGAAAAACGAATTCTTAAAAATGCAGTTCAACTCTATCGTCATTCAACTGAAGCCAGCCGTGGCTCGGCTTCACAAGAAAG
head apul.contigs.fasta
>tig00000004 len=43693 reads=3 class=contig suggestRepeat=no suggestBubble=yes suggestCircular=no trim=0-43693
TACAATTTTAGAACACGGGACCAGCTTAGCATAATAGCTTCACCTTTCGTCTATCTAACTCTAGGAAGTTTTAATTTTTTCAAGTATTATAAAGGGCTCC
GTCGACTGTCAAAGATTTGCCTTTTCAAGCTCCAATGGAAACTGTAAAAGTTGCGTATTTTTACGAGCTTGAAACGCATCTTGGTATGCCCGATATCAAG
TCGAAATTAATATTGAGATAATCCTTTGGCCTTCTTCTATCAACATCTGAAATAAAAATCTGGGCAGTTTGAACGCGCTCTATCAAACATAACAGATTTG
AAGGTAGGTGATAACTTATTTGCATAATCTACGTTAACAAAAAGTCTATTTATAGAATGACTACTCGGCATATTTCTAACAGTGGTACTTCAGATACGTT
TTGATGGACTTATTATTCTGTCGTTTGTATTGTTTTCTTCAATTATTTAGCCTTAATAATTCCAAATAATAAAGAAATAAGGAAAGTCTTTGGTGTAAGT
CACACTCAAAAGGTGAGTTTCAACAGTTACTGAACACCCTTACGTATTAAACAGTCATTTCAATTTCCAGATTCTAACAGAAAATGTCAAATCGTTGTTT
TATAGTAGAAATCCATCTTCAAAAGTTATTCCCCGCTTATGCAGGCTTGATTCTCGCGGCTCTTTCCAGCTCGGTTTACAATATAAGACACCGGTGCAGA
TACCATTGAACTTGTAAACAATGTCACGCAAATTAAACTGTACTTCAATTTGCAAGCCATACAGCTTTAAGTCAGGTCTTTATTGAACTTTCTAAGTCAA
GGTTGGGGAATATAAAGATATTTTATTACCAGTATATTTTCGGTGAAAATTACAACGGATACATGTTATGGGCCTGTTCTTTAAACTCAGTTACATACAT
The unassembled file contains the reads and low-coverage contigs which couldn’t be incorporated into the primary assembly. The contigs file contains the full assembly, including unique, repetitive, and bubble elements. What does this mean? Unsure, but the header line provides metadata on each sequence.
len
Length of the sequence, in bp.
reads
Number of reads used to form the contig.
class
Type of sequence. Unassembled sequences are primarily low-coverage sequences spanned by a single read.
suggestRepeat
If yes, sequence was detected as a repeat based on graph topology or read overlaps to other sequences.
suggestBubble
If yes, sequence is likely a bubble based on potential placement within longer sequences.
suggestCircular
If yes, sequence is likely circular. The fasta line will have a trim=X-Y to indicate the non-redundant coordinates
If we are looking at this header: >tig00000004 len=43693 reads=3 class=contig suggestRepeat=no suggestBubble=yes suggestCircular=no trim=0-43693, the length is the contig is 43693 bp, 3 reads were used to form the contig, class is a contig, no repeats used to form the contig, the sequence is likely a bubble based on placement within longer sequences, the sequence is not circular, and the entire contig is non-redundant. Not sure what bubble means…
Because Canu didn’t trim or correct the Hifi reads, I may need to use a different assembly tool. I’m going to try Hifiasm, which is a fast haplotype-resolved de novo assembler designed for PacBio HiFi reads. According to Hifiasm github, here are some good reasons to use Hifiasm:
-
Hifiasm delivers high-quality telomere-to-telomere assemblies. It tends to generate longer contigs and resolve more segmental duplications than other assemblers.
-
Hifiasm can purge duplications between haplotigs without relying on third-party tools such as purge_dups. Hifiasm does not need polishing tools like pilon or racon, either. This simplifies the assembly pipeline and saves running time.
-
Hifiasm is fast. It can assemble a human genome in half a day and assemble a ~30Gb redwood genome in three days. No genome is too large for hifiasm.
-
Hifiasm is trivial to install and easy to use. It does not required Python, R or C++11 compilers, and can be compiled into a single executable. The default setting works well with a variety of genomes.
If I use this tool, I may not need to pilon to polish the assembly. This tool isn’t on the server, so I’ll need to create a conda environment and install the package.
cd /data/putnamlab/conda
mkdir hifiasm
cd hifiasm
module load Miniconda3/4.9.2
conda create --prefix /data/putnamlab/conda/hifiasm
conda activate /data/putnamlab/conda/hifiasm
conda install -c bioconda hifiasm
Once this package is installed, run code for hifiasm assembly. In the scripts folder: nano hifiasm.sh
#!/bin/bash -i
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/hifiasm
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting assembly with hifiasm" $(date)
hifiasm -o apul.hifiasm m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq
echo "Assembly with hifiasm complete!" $(date)
conda deactivate
Submitted batch job 300534
20240213
Even though the reads were not trimmed or corrected with Canu, I am going to run Busco on the output. This will provide information about how well the genome was assembled and its completeness based on evolutionarily informed expectations of gene content from near-universal single-copy orthologs. Danielle and Kevin have both run BUSCO before and used similar scripts but I think I’ll adapt mine a little to fit my needs and personal preferences for code.
From the Busco user manual, the mandatory parameters are -i, which defines the input fasta file and -m, which sets the assessment mode (in our case, genome). Some recommended parameters incude l (specify busco lineage dataset; in our case, metazoans), c (specify number of cores to use), and -o (assigns specific label to output).
In /data/putnamlab/shared/busco/scripts, the script busco_init.sh has information about the modules to load and in what order. Both Danielle and Kevin sourced this file specifically in their code, but I will probably just copy and paste the modules. In the same folder, they also used busco-config.ini as input for the --config flag in busco, which provides a config file as an alternative to command line parameters. I am not going to use this config file (yet), as Danielle and Kevin were assembling transcriptomes and I’m not sure what the specifics of the file are (or what they should be for genomes). In /data/putnamlab/shared/busco/downloads/lineages/metazoa_odb10, there is information about the metazoan database.
In /data/putnamlab/jillashey/Apul_Genome/assembly/scripts, nano busco_canu.sh
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BUSCO/5.2.2-foss-2020b
module load BLAST+/2.11.0-gompi-2020b
module load AUGUSTUS/3.4.0-foss-2020b
module load SEPP/4.4.0-foss-2020b
module load prodigal/2.6.3-GCCcore-10.2.0
module load HMMER/3.3.2-gompi-2020b
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Begin busco on canu-assembled fasta" $(date)
busco -i apul.contigs.fasta -m genome -l /data/putnamlab/shared/busco/downloads/lineages/metazoa_odb10 -c 15 -o apul.busco.canu
echo "busco complete for canu-assembled fasta" $(date)
Submitted batch job 301588. This failed and gave me some errors. This one seemed to have been the fatal one: Message: BatchFatalError(AttributeError("'NoneType' object has no attribute 'remove_tmp_files'")). Danielle ran into a similar error in her busco code so I am going to try to set the --config file as "$EBROOTBUSCO/config/config.ini".
In the script, nano busco_canu.sh
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
#module load BUSCO/5.2.2-foss-2020b
#module load BLAST+/2.11.0-gompi-2020b
#module load AUGUSTUS/3.4.0-foss-2020b
#module load SEPP/4.4.0-foss-2020b
#module load prodigal/2.6.3-GCCcore-10.2.0
#module load HMMER/3.3.2-gompi-2020b
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Begin busco on canu-assembled fasta" $(date)
source "/data/putnamlab/shared/busco/scripts/busco_init.sh" # sets up the modules required for this in the right order
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 15 --long -i apul.contigs.fasta -m genome -l /data/putnamlab/shared/busco/downloads/lineages/metazoa_odb10 -o apul.busco.canu
echo "busco complete for canu-assembled fasta" $(date)
Submitted batch job 301594. Failed, same error as before. Going to try copying Kevin and Danielle code directly, even though its a little messy and confusing with paths.
In the script, nano busco_canu.sh
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Begin busco on canu-assembled fasta" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.contigs.fasta" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.busco.canu -m genome
echo "busco complete for canu-assembled fasta" $(date)
Submitted batch job 301599. This appears to have worked! Took about an hour to run. This is the primary result in the out file:
# BUSCO version is: 5.2.2
# The lineage dataset is: metazoa_odb10 (Creation date: 2024-01-08, number of genomes: 65, number of BUSCOs: 954)
# Summarized benchmarking in BUSCO notation for file /data/putnamlab/jillashey/Apul_Genome/assembly/data/apul.contigs.fasta
# BUSCO was run in mode: genome
# Gene predictor used: metaeuk
***** Results: *****
C:94.4%[S:9.4%,D:85.0%],F:2.7%,M:2.9%,n:954
901 Complete BUSCOs (C)
90 Complete and single-copy BUSCOs (S)
811 Complete and duplicated BUSCOs (D)
26 Fragmented BUSCOs (F)
27 Missing BUSCOs (M)
954 Total BUSCO groups searched
We have 94.4% completeness with this assembly but 85% complete and duplicated BUSCOs. The busco manual says this on high levels of duplication: “BUSCO completeness results make sense only in the context of the biology of your organism. You have to understand whether missing or duplicated genes are of biological or technical origin. For instance, a high level of duplication may be explained by a recent whole duplication event (biological) or a chimeric assembly of haplotypes (technical). Transcriptomes and protein sets that are not filtered for isoforms will lead to a high proportion of duplicates. Therefore you should filter them before a BUSCO analysis”.
Danielle also got a high number (78.9%) of duplicated BUSCOs in her de novo transcriptome of Apulchra, but Kevin got much less duplication (6.9%) in his Past transcriptome assembly. I need to ask Danielle if she ended up using her Trinity results (which had a high duplication percentage) for her alignment for Apul. I also need to ask her if she thinks the high duplication percentage is biologically meaningful.
Might be worth running HiFiAdapter Filt
20240215
Last night, the hifiasm job failed after almost 2 days but the email says PREEMPTED, ExitCode0. Two minutes after the job failed, job 300534 started again on the server and it says its a hifiasm job…I did not start this job myself, not sure what happened. Looking on the server now, hifiasm is running but has only been running for about 18 hours (as of 2pm today). It’s the same job number though which is strange.
20240220
Hifiasm job is still running after ~5 days. In the meantime, I’m going to run HiFiAdapterFilt, which is an adapter filtering command for PacBio HiFi data. On the github page, it says that the tool converts .bam to .fastq and removes reads with remnant PacBio adapter sequences. Required dependencies are BamTools and BLAST+; optional dependencies are NCBI FCS Adaptor and pigz. It looks like I’ll need to use the original bam file instead of the converted fastq file.
The github says I should add the script and database to my path using:
export PATH=$PATH:[PATH TO HiFiAdapterFilt]
export PATH=$PATH:[PATH TO HiFiAdapterFilt]/DB
I will do this in the script for the adapter filt code that I write myself. In the scripts folder, make a folder for hifi information
mkdir HiFiAdapterFilt
cd HiFiAdapterFilt
I need to make a script for the hifi adapter code. In the scripts/HiFiAdapterFilt folder, I copy and pasted the linked code into hifiadapterfilt.sh. Make a folder for pacbio databases and copy in the db information from the github.
mkdir DB
cd DB
nano pacbio_vectors_db
>gnl|uv|NGB00972.1:1-45 Pacific Biosciences Blunt Adapter
ATCTCTCTCTTTTCCTCCTCCTCCGTTGTTGTTGTTGAGAGAGAT
>gnl|uv|NGB00973.1:1-35 Pacific Biosciences C2 Primer
AAAAAAAAAAAAAAAAAATTAACGGAGGAGGAGGA
In the scripts/HiFiAdapterFilt folder: nano hifiadapterfilt_JA.sh
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts/HiFiAdapterFilt
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load GCCcore/11.3.0 # need this to resolve conflicts between GCCcore/8.3.0 and loaded GCCcore/11.3.0
module load BamTools/2.5.1-GCC-8.3.0
module load BLAST+/2.9.0-iimpi-2019b
cd /data/putnamlab/jillashey/Apul_Genome/assembly/scripts/HiFiAdapterFilt
echo "Setting paths" $(date)
export PATH=$PATH:[/data/putnamlab/jillashey/Apul_Genome/assembly/scripts/HiFiAdapterFilt] # path to original script
export PATH=$PATH:[/data/putnamlab/jillashey/Apul_Genome/assembly/scripts/HiFiAdapterFilt]/DB # path to db info
echo "Paths set, starting adapter filtering" $(date)
bash hifiadapterfilt.sh -p /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029 -l 44 -m 97 -o /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Completing adapter filtering" $(date)
The -l and -m refer to the minimum length of adapter match to remove and the minumum percent match of adapter to remove, respectively. I left them as the default settings for now. Submitted batch job 303636. Giving me this error:
hifiadapterfilt.sh: line 63: /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029.temp_file_list: Permission denied
cat: /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029.bam.temp_file_list: No such file or directory
cat: /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029.bam.temp_file_list: No such file or directory
Giving me permission denied to write in the folder? I’ll sym link the bam file to my data folder
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
ln -s /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029.bam
Editing script so that the prefix is connecting with the sym linked file. Submitted batch job 303637. Still giving me the same error. Hollie may need to give me permission to write and access files in that specific folder.
20240221
Probably need to run haplomerger2, which is installed on the server already.
I’m also looking at the Canu FAQs to see if there is any info about using PacBio HiFi reads. Under the question “What parameters should I use for my reads?”, they have this info:
The defaults for -pacbio-hifi should work on this data. There is still some variation in data quality between samples. If you have poor continuity, it may be because the data is lower quality than expected. Canu will try to auto-adjust the error thresholds for this (which will be included in the report). If that still doesn’t give a good assembly, try running the assembly with -untrimmed. You will likely get a genome size larger than you expect, due to separation of alleles. See My genome size and assembly size are different, help! for details on how to remove this duplication.
When I look at the question “My genome size and assembly size are different, help!”, it says that this difference could be due to a heterozygous genome where the assembly separated some loci or the previous estimate is incorrect. They recommended running BUSCO to check completeness of the assembly (which I already did) and using purge_dups to remove duplication. I will look into this.
Next steps
- Run Canu with
-untrimmedoption - Run purge_dups - targets the removal of duplicated sequences to enhance overall quality of assembly
- Run haplomerger - merges haplotypes to addresws heterozygosity
In the scripts folder: nano canu_untrimmed.sh
#!/bin/bash
#SBATCH -t 500:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load canu/2.2-GCCcore-11.2.0
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
#echo "Unzip paco-bio fastq file" $(date)
#gunzip m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq.gz
echo "Starting assembly w/ untrimmed flag" $(date)
canu -p apul.canu.untrimmed -d /data/putnamlab/jillashey/Apul_Genome/assembly/data genomeSize=475m -raw -pacbio-hifi m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq
echo "Canu assembly complete" $(date)
Submitted batch job 303660
On the purge_dups github, they say to install using the following:
git clone https://github.com/dfguan/purge_dups.git
cd purge_dups/src && make
# only needed if running run_purge_dups.py
git clone https://github.com/dfguan/runner.git
cd runner && python3 setup.py install --user
Cloned both into the assembly folder (ie /data/putnamlab/jillashey/Apul_Genome/assembly/).
First, use pd_config.py to generate a configuration file. Here’s possible usage:
usage: pd_config.py [-h] [-s SRF] [-l LOCD] [-n FN] [--version] ref pbfofn
generate a configuration file in json format
positional arguments:
ref reference file in fasta/fasta.gz format
pbfofn list of pacbio file in fastq/fasta/fastq.gz/fasta.gz format (one absolute file path per line)
optional arguments:
-h, --help show this help message and exit
-s SRF, --srfofn SRF list of short reads files in fastq/fastq.gz format (one record per line, the
record is a tab splitted line of abosulte file path
plus trimmed bases, refer to
https://github.com/dfguan/KMC) [NONE]
-l LOCD, --localdir LOCD
local directory to keep the reference and lists of the
pacbio, short reads files [.]
-n FN, --name FN output config file name [config.json]
--version show program's version number and exit
# Example
./scripts/pd_config.py -l iHelSar1.pri -s 10x.fofn -n config.iHelSar1.PB.asm1.json ~/vgp/release/insects/iHelSar1/iHlSar1.PB.asm1/iHelSar1.PB.asm1.fa.gz pb.fofn
I need to make a list of the pacbio files that I’ll use in the script and put it in a file called pb.fofn.
for filename in *.fastq; do echo $PWD/$filename; done > pb.fofn
In my scripts folder: nano pd_config_apul.py
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Python/3.9.6-GCCcore-11.2.0 # do i need python?
echo "Making config file for purge dups scripts" $(date)
/data/putnamlab/jillashey/Apul_Genome/assembly/purge_dups/scripts/pd_config.py -l /data/putnamlab/jillashey/Apul_Genome/assembly/data -n config.apul.canu.json /data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq /data/putnamlab/jillashey/Apul_Genome/assembly/data/pb.fofn
echo "Config file complete" $(date)
Submitted batch job 303661. Ran in 1 second. Got this in the error file:
cp: ‘/data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq’ and ‘/data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq’ are the same file
cp: ‘/data/putnamlab/jillashey/Apul_Genome/assembly/data/pb.fofn’ and ‘/data/putnamlab/jillashey/Apul_Genome/assembly/data/pb.fofn’ are the same file
But it did generate a config file, which looks like this:
{
"cc": {
"fofn": "/data/putnamlab/jillashey/Apul_Genome/assembly/data/pb.fofn",
"isdip": 1,
"core": 12,
"mem": 20000,
"queue": "normal",
"mnmp_opt": "",
"bwa_opt": "",
"ispb": 1,
"skip": 0
},
"sa": {
"core": 12,
"mem": 10000,
"queue": "normal"
},
"busco": {
"core": 12,
"mem": 20000,
"queue": "long",
"skip": 0,
"lineage": "mammalia",
"prefix": "m84100_240128_024355_s2.hifi_reads.bc1029.fastq_purged",
"tmpdir": "busco_tmp"
},
"pd": {
"mem": 20000,
"queue": "normal"
},
"gs": {
"mem": 10000,
"oe": 1
},
"kcp": {
"core": 12,
"mem": 30000,
"fofn": "",
"prefix": "m84100_240128_024355_s2.hifi_reads.bc1029.fastq_purged_kcm",
"tmpdir": "kcp_tmp",
"skip": 1
},
"ref": "/data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq",
"out_dir": "m84100_240128_024355_s2.hifi_reads.bc1029.fastq"
}
My config file looks basically the same as the example one on the github. Manually edited the config file so that the out_dir was /data/putnamlab/jillashey/Apul_Genome/assembly/data/. Now the purging can begin using run_purge_dups.py. Here’s possible usage:
usage: run_purge_dups.py [-h] [-p PLTFM] [-w WAIT] [-r RETRIES] [--version]
config bin_dir spid
purge_dups wrapper
positional arguments:
config configuration file
bin_dir directory of purge_dups executable files
spid species identifier
optional arguments:
-h, --help show this help message and exit
-p PLTFM, --platform PLTFM
workload management platform, input bash if you want to run locally
-w WAIT, --wait WAIT <int> seconds sleep intervals
-r RETRIES, --retries RETRIES
maximum number of retries
--version show program's version number and exit
# Example
python scripts/run_purge_dups.py config.iHelSar1.json src iHelSar1
In my scripts folder: nano run_purge_dups_apul.py
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Python/3.9.6-GCCcore-11.2.0 # do i need python?
echo "Starting to purge duplications" $(date)
/data/putnamlab/jillashey/Apul_Genome/assembly/purge_dups/scripts/run_purge_dups.py /data/putnamlab/jillashey/Apul_Genome/assembly/scripts/config.apul.canu.json /data/putnamlab/jillashey/Apul_Genome/assembly/purge_dups/src apul
echo "Duplication purge complete" $(date)
Submitted batch job 303662. Failed immediately with this error:
Traceback (most recent call last):
File "/data/putnamlab/jillashey/Apul_Genome/assembly/purge_dups/scripts/run_purge_dups.py", line 3, in <module>
from runner.manager import manager
ModuleNotFoundError: No module named 'runner'
I installed runner but the code is not seeing it…where am I supposed to put it? inside of the purge_dups github? Okay going to move runner folder inside of the purge_dups folder.
cd /data/putnamlab/jillashey/Apul_Genome/assembly
mv runner/ purge_dups/scripts/
Submitting job again, Submitted batch job 303663. Got the same error. In the run_purge_dups.py script itself, the first few lines are:
#!/usr/bin/env python3
from runner.manager import manager
from runner.hpc import hpc
from multiprocessing import Process, Pool
import sys, os, json
import argparse
So it isn’t seeing that the runner module is there. This issue and this issue on the github were reported but never really answered in a clear way. Will have to look into this more.
In other news, the canu script finished running but looks like it failed. This is the bottom of the error message:
ERROR:
ERROR: Failed with exit code 139. (rc=35584)
ERROR:
ABORT:
ABORT: canu 2.2
ABORT: Don't panic, but a mostly harmless error occurred and Canu stopped.
ABORT: Try restarting. If that doesn't work, ask for help.
ABORT:
ABORT: failed to configure the overlap store.
ABORT:
ABORT: Disk space available: 8477.134 GB
ABORT:
ABORT: Last 50 lines of the relevant log file (unitigging/apul.canu.untrimmed.ovlStore.config.err):
ABORT:
ABORT:
ABORT: Finding number of overlaps per read and per file.
ABORT:
ABORT: Moverlaps
ABORT: ------------ ----------------------------------------
ABORT:
ABORT: Failed with 'Segmentation fault'; backtrace (libbacktrace):
ABORT:
Unsure what it means…
20240301
BIG NEWS!!!!! This week, a paper came out that assembled and annotated the Orbicella faveolata genome using PacBio HiFi reads (Young et al. 2024)!!!!!!! The github for this paper has a detailed pipeline for how the genome was put together. Since I am also using HiFi reads, I will be following their methodology! I am using this pipeline starting at line 260.
I changed the file from bam to fastq, but now I need to change it to fasta with seqtk. In the scripts folder: nano seqtk.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load seqtk/1.3-GCC-9.3.0
echo "Convert PacBio fastq file to fasta file" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
seqtk seq -a m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq > m84100_240128_024355_s2.hifi_reads.bc1029.fasta
echo "Fastq to fasta complete! Summarize read lengths" $(date)
awk '/^>/{printf("%s\t",substr($0,2));next;} {print length}' m84100_240128_024355_s2.hifi_reads.bc1029.fasta > rr_read_lengths.txt
echo "Read length summary complete" $(date)
Submitted batch job 304257.
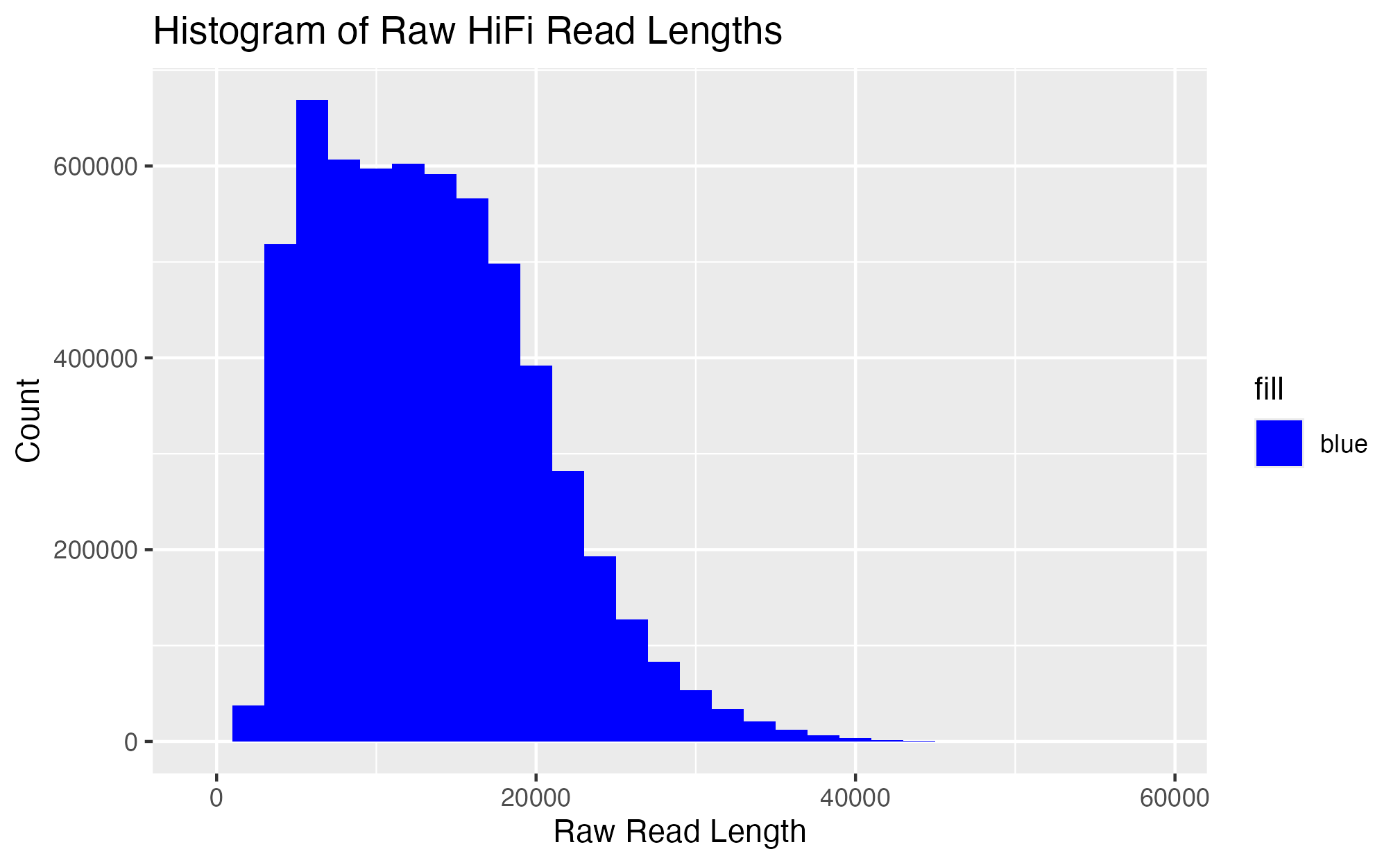
In R, I looked at the data to quantify length for each read. See code here.
```{r, echo=F} read.table(file = “../data/rr_read_lengths.txt”, header = F) %>% dplyr::rename(“hifi_read_name” = 1, “length” = 2) -> hifi_read_length nrow(hifi_read_length) # 5,898,386 total reads mean(hifi_read_length$length) # mean length of reads is 13,424.64 sum(hifi_read_length$length) #length sum 79,183,709,778. Will need this for the NCBI submission
Make histogram for read bins from raw hifi data
```{r, echo = F}
ggplot(data = hifi_read_length,
aes(x = length, fill = "blue")) +
geom_histogram(binwidth = 2000) +
labs(x = "Raw Read Length", y = "Count", title = "Histogram of Raw HiFi Read Lengths") +
scale_fill_manual(values = c("blue")) +
scale_y_continuous(labels = function(x) format(x, scientific = FALSE))
20240303
My next step is to remove any contaminant reads from the raw hifi reads. From Young et al. 2024: “Raw HiFi reads first underwent a contamination screening, following the methodology in [68], using BLASTn [32, 68] against the assembled mitochondrial O. faveolata genome and the following databases: common eukaryote contaminant sequences (ftp.ncbi.nlm.nih. gov/pub/kitts/contam_in_euks.fa.gz), NCBI viral (ref_ viruses_rep_genomes) and prokaryote (ref_prok_rep_ genomes) representative genome sets”.
I tried to run the update_blastdb.pl script (included in the blast program) with the BLAST+/2.13.0-gompi-2022a module but I got this error:
Can't locate Archive/Tar.pm in @INC (@INC contains: /usr/local/lib64/perl5 /usr/local/share/perl5 /usr/lib64/perl5/vendor_perl /usr/share/perl5/vendor_perl /usr/lib64/perl5 /usr/share/perl5 .) at /opt/software/BLAST+/2.13.0-gompi-2022a/bin/update_blastdb.pl line 41.
BEGIN failed--compilation aborted at /opt/software/BLAST+/2.13.0-gompi-2022a/bin/update_blastdb.pl line 41.
Not sure what this means…will email Kevin Bryan to ask about it, as I don’t want to mess with anything on the installed modules. I did download the contam_in_euks.fa.gz db to my computer so I’m going to copy it to Andromeda. This file is considerably smaller than the viral or prok dbs.
Make a database folder in the Apul genome folder.
cd /data/putnamlab/jillashey/Apul_Genome
mkdir dbs
cd dbs
zgrep -c ">" contam_in_euks.fa
3554
Now I ran run a script that blasts the pacbio fasta against these sequences. In /data/putnamlab/jillashey/Apul_Genome/assembly/scripts, nano blast_contam_euk.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
echo "BLASTing hifi fasta against eukaryote contaminant sequences" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -subject /data/putnamlab/jillashey/Apul_Genome/dbs/contam_in_euks.fa -task megablast -outfmt 6 -evalue 4 -perc_identity 90 -num_threads 15 -out contaminant_hits_euks_rr.txt
echo "BLAST complete, remove contaminant seqs from hifi fasta" $(date)
awk '{ if( ($4 >= 50 && $4 <= 99 && $3 >=98 ) ||
($4 >= 100 && $4 <= 199 && $3 >= 94 ) ||
($4 >= 200 && $3 >= 90) ) {print $0}
}' contaminant_hits_euks_rr.txt > contaminants_pass_filter_euks_rr.txt
echo "Contaminant seqs removed from hifi fasta" $(date)
Submitted batch job 304389. Finished in about 2.5 hours. Looked at the output in R (code here).
20240304
Emailed Kevin Bryan this morning and asked if he knew anything about why the update_blastdb.pl wasn’t working. Still waiting to hear back from him.
Kevin Bryan also emailed me this morning about my hifiasm job that has been running for 18 days and said: “This job has been running for 18 days. I just took a look at it and it appears you didn’t specify -t $SLURM_CPUS_ON_NODE (and also #SBATCH --exclusive) to make use of all of the CPU cores on the node. You might want to consider re-submitting this job with those parameters. Because the nodes generally have 36 cores, it should be able to catch up to where it is now in a little over half a day, assuming perfect scaling.”
I need to add those parameters into the hifiasm code, so cancelling the 300534 job. In /data/putnamlab/jillashey/Apul_Genome/assembly/scripts, nano hifiasm.sh:
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/hifiasm
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting assembly with hifiasm" $(date)
hifiasm -o apul.hifiasm m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq -t 36
echo "Assembly with hifiasm complete!" $(date)
conda deactivate
Submitted batch job 304463
20240304
Response from Kevin Bryan about viral and prok blast databases: “Ok, I downloaded those databases, and actually consolidated the rest of them into /data/shared/ncbi-db/, under which is a directory for today, and then there will be a new one next Sunday and following Sundays. There’s a file /data/shared/ncbi-db/.ncbirc that gets updated to point to the current directory, which the blast* tools will automatically pick up, so you can just do -db ref_prok_rep_ genomes, for example.
For other tools that can read the ncbi databases, you can use blastdb_path -db ref_viruses_rep_genomes to get the path, although for some reason with the nr database you need to specify -dbtype prot, i.e., blastdb_path -db nr -dbtype prot.
The reason for the extra complication is because otherwise a job that runs while the database is being updated may fail or return strange results. The dated directories should resolve this issue. Note that Unity blast-plus modules work in a similar way with a different path, /datasets/bio/ncbi-db.”
Amazing! Now I can move forward with the blasting against viral and prok genomes. In the scripts folder: nano blastn_viral.sh
#!/bin/bash
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Blasting hifi reads against viral genomes to look for contaminants" $(date)
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -db ref_viruses_rep_genomes -outfmt 6 -evalue 1e-4 -perc_identity 90 -out viral_contaminant_hits_rr.txt
echo "Blast complete!" $(date)
Submitted batch job 304500
In the scripts folder: nano blastn_prok.sh
#!/bin/bash
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Blasting hifi reads against prokaryote genomes to look for contaminants" $(date)
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -db ref_prok_rep_genomes -outfmt 6 -evalue 1e-4 -perc_identity 90 -out prok_contaminant_hits_rr.txt
echo "Blast complete!" $(date)
Submitted batch job 304502
20240306
Making list of all software programs that Young et al. 2024 used and if they are on Andromeda
- blastn
- On Andromeda? YES
- Meryl
- On Andromeda? NO
- Genome-Scope2
- On Andromeda? NO
- Hifiasm
- On Andromeda? NO but I added it to
putnamlabvia conda
- On Andromeda? NO but I added it to
- Quast
- On Andromeda? YES
- Busco
- On Andromeda? YES
- Merqury
- On Andromeda? NO
- RepeatModeler2
- On Andromeda? NO
- Repeat-Masker
- On Andromeda? YES
- TeloScafs
- On Andromeda? NO
- PASA
- On Andromeda? NO
- funnannotate
- On Andromeda? NO
- Augustus
- On Andromeda? YES
- GeneMark-ES/ET
- On Andromeda? YES but only GeneMark-ET
- snap
- On Andromeda? YES
- glimmerhmm
- On Andromeda? NO
- Evidence Modeler
- On Andromeda? NO
- tRNAscan-SE
- On Andromeda? YES
- Trinity
- On Andromeda? Yes
- InterproScan
- On Andromeda? YES
20240311
Hifiasm (with unfiltered reads) finished running over the weekend and the prok blast script preemptively ended and then restarted in the early hours of this morning. I think this might be because I am not making use of all cores on the node (similar to my earlier hifiasm script). I cancelled the prok blast job (304502) and edited the script so that it includes the flag -num_threads 36. Submitted batch job 305351
It created many files:
-rw-r--r--. 1 jillashey 19G Mar 7 23:58 apul.hifiasm.ec.bin
-rw-r--r--. 1 jillashey 47G Mar 8 00:08 apul.hifiasm.ovlp.source.bin
-rw-r--r--. 1 jillashey 17G Mar 8 00:12 apul.hifiasm.ovlp.reverse.bin
-rw-r--r--. 1 jillashey 1.2G Mar 8 01:37 apul.hifiasm.bp.r_utg.gfa
-rw-r--r--. 1 jillashey 21M Mar 8 01:37 apul.hifiasm.bp.r_utg.noseq.gfa
-rw-r--r--. 1 jillashey 8.6M Mar 8 01:41 apul.hifiasm.bp.r_utg.lowQ.bed
-rw-r--r--. 1 jillashey 1.1G Mar 8 01:42 apul.hifiasm.bp.p_utg.gfa
-rw-r--r--. 1 jillashey 21M Mar 8 01:42 apul.hifiasm.bp.p_utg.noseq.gfa
-rw-r--r--. 1 jillashey 8.2M Mar 8 01:46 apul.hifiasm.bp.p_utg.lowQ.bed
-rw-r--r--. 1 jillashey 506M Mar 8 01:47 apul.hifiasm.bp.p_ctg.gfa
-rw-r--r--. 1 jillashey 11M Mar 8 01:47 apul.hifiasm.bp.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 2.0M Mar 8 01:49 apul.hifiasm.bp.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 469M Mar 8 01:50 apul.hifiasm.bp.hap1.p_ctg.gfa
-rw-r--r--. 1 jillashey 9.9M Mar 8 01:50 apul.hifiasm.bp.hap1.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 2.0M Mar 8 01:52 apul.hifiasm.bp.hap1.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 468M Mar 8 01:52 apul.hifiasm.bp.hap2.p_ctg.gfa
-rw-r--r--. 1 jillashey 9.9M Mar 8 01:52 apul.hifiasm.bp.hap2.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 1.9M Mar 8 01:54 apul.hifiasm.bp.hap2.p_ctg.lowQ.bed
This page gives a brief overview of the hifiasm output files, which is super helpful. It generates the assembly graphs in Graphical Fragment Assembly (GFA) format.
- prefix.r_utg.gfa: haplotype-resolved raw unitig graph. This graph keeps all haplotype information.
- A unitig is a portion of a contig. It is a nondisputed and assembled group of fragments. A contiguous sequence of ordered unitigs is a contig, and a single unitig can be in multiple contigs.
- prefix.p_utg.gfa: haplotype-resolved processed unitig graph without small bubbles. Small bubbles might be caused by somatic mutations or noise in data, which are not the real haplotype information. Hifiasm automatically pops such small bubbles based on coverage. The option –hom-cov affects the result. See homozygous coverage setting for more details. In addition, the option -p forcedly pops bubbles.
- Confused about the bubbles, but it looks like a medium level (what is a “medium” level”?) of heterozygosity will result in bubbles (see image in this post)
- Homozygous coverage refers to coverage threshold for homozygous reads. Hifiasm prints it as:
[M::purge_dups] homozygous read coverage threshold: X. If it is not around homozygous coverage, the final assembly might be either too large or too small.
- prefix.p_ctg.gfa: assembly graph of primary contigs. This graph includes a complete assembly with long stretches of phased blocks.
- From my understanding based on this post discussing concepts in phased assemblies, a phased assembly identifies different alleles
- prefix.a_ctg.gfa: assembly graph of alternate contigs. This graph consists of all contigs that are discarded in primary contig graph.
- There were none of these files in my output. Does this mean all contigs created were used in the final assembly?
- prefix.hap*.p_ctg.gfa: phased contig graph. This graph keeps the phased contigs for haplotype 1 and haplotype 2.
I believe this file (apul.hifiasm.bp.p_ctg.gfa) contains the sequence information for the assembled contigs. zgrep -c "S" apul.hifiasm.bp.p_ctg.gfa showed that there were 188 Segments, or continuous sequences, in this assembly, meaning there are 188 contigs (to my understanding). This file (apul.hifiasm.bp.p_ctg.noseq.gfa) contains information about the reads used to construct the contigs in a plain text format.
head apul.hifiasm.bp.p_ctg.noseq.gfa
S ptg000001l * LN:i:21642937 rd:i:82
A ptg000001l 0 + m84100_240128_024355_s2/165481517/ccs 0 25806 id:i:5084336 HG:A:a
A ptg000001l 6512 + m84100_240128_024355_s2/221910786/ccs 0 21626 id:i:5609370 HG:A:a
A ptg000001l 7287 - m84100_240128_024355_s2/128778986/ccs 0 24026 id:i:2691352 HG:A:a
A ptg000001l 7493 + m84100_240128_024355_s2/262476615/ccs 0 30540 id:i:287120 HG:A:a
A ptg000001l 15042 + m84100_240128_024355_s2/191824085/ccs 0 27619 id:i:2783058 HG:A:a
A ptg000001l 16616 - m84100_240128_024355_s2/29886096/ccs 0 28664 id:i:2336674 HG:A:a
A ptg000001l 17527 - m84100_240128_024355_s2/37553051/ccs 0 31883 id:i:2336554 HG:A:a
A ptg000001l 21104 - m84100_240128_024355_s2/242419829/ccs 0 28551 id:i:5120251 HG:A:a
A ptg000001l 21788 + m84100_240128_024355_s2/266536351/ccs 0 27994 id:i:5577462 HG:A:a
The S line is the Segment and it acts as a header for the the contig. LN:i: is the segment length (in this case, 21,642,937 bp). The rd:i: is the read coverage, calculated by the reads coming from the same contig (in this case, read coverage is 82, which is high). The A lines provide information about the sequences that make up the contig. Here’s what each column means
- Column 1: should always be A
- Column 2: contig name
- Column 3: contig start coordinate of subregion constructed by read
- Column 4: read strand (+ or -)
- Column 5: read name
- Column 6: read start coordinate of subregion which is used to construct contig
- Column 7: read end coordinate of subregion which is used to construct contig
- Column 8: read ID
- Column 9: haplotype status of read.
HG:A:a,HG:A:p, andHG:A:mindicate that the read is non-binnable (ie heterozygous), father/hap1 specific, or mother/hap2 specific.
If I’m interpreting this correctly, it looks like most of the reads in the first contig are heterozygous.
The error file (slurm-304463.error) for this script contains the histogram of the kmers. It has 4 iterations of kmer histograms with some sort of analysis in between the histograms. Here’s what the last histogram looks like:
[M::ha_analyze_count] lowest: count[9] = 2315
[M::ha_analyze_count] highest: count[83] = 417609
[M::ha_hist_line] 1: ****************************************************************************************************> 1732549
[M::ha_hist_line] 2: ************* 53793
[M::ha_hist_line] 3: **** 16969
[M::ha_hist_line] 4: ** 9184
[M::ha_hist_line] 5: * 5507
[M::ha_hist_line] 6: * 4361
[M::ha_hist_line] 7: * 3389
[M::ha_hist_line] 8: * 2959
[M::ha_hist_line] 9: * 2315
[M::ha_hist_line] 10: * 2369
[M::ha_hist_line] 11: 2037
[M::ha_hist_line] 12: 1889
[M::ha_hist_line] 13: 1724
[M::ha_hist_line] 14: 1729
[M::ha_hist_line] 15: 1721
[M::ha_hist_line] 16: 1641
[M::ha_hist_line] 17: 1530
[M::ha_hist_line] 18: 1687
[M::ha_hist_line] 19: 1389
[M::ha_hist_line] 20: 1341
[M::ha_hist_line] 21: 1370
[M::ha_hist_line] 22: 1269
[M::ha_hist_line] 23: 1249
[M::ha_hist_line] 24: 1329
[M::ha_hist_line] 25: 1327
[M::ha_hist_line] 26: 1317
[M::ha_hist_line] 27: 1274
[M::ha_hist_line] 28: 1370
[M::ha_hist_line] 29: 1495
[M::ha_hist_line] 30: 1468
[M::ha_hist_line] 31: 1677
[M::ha_hist_line] 32: 1625
[M::ha_hist_line] 33: 1729
[M::ha_hist_line] 34: 1697
[M::ha_hist_line] 35: 1825
[M::ha_hist_line] 36: 1919
[M::ha_hist_line] 37: 1966
[M::ha_hist_line] 38: 2066
[M::ha_hist_line] 39: * 2101
[M::ha_hist_line] 40: * 2195
[M::ha_hist_line] 41: * 2119
[M::ha_hist_line] 42: * 2100
[M::ha_hist_line] 43: * 2325
[M::ha_hist_line] 44: * 2644
[M::ha_hist_line] 45: * 2807
[M::ha_hist_line] 46: * 3080
[M::ha_hist_line] 47: * 3289
[M::ha_hist_line] 48: * 3661
[M::ha_hist_line] 49: * 3984
[M::ha_hist_line] 50: * 4856
[M::ha_hist_line] 51: * 5391
[M::ha_hist_line] 52: ** 6627
[M::ha_hist_line] 53: ** 7648
[M::ha_hist_line] 54: ** 9319
[M::ha_hist_line] 55: *** 11051
[M::ha_hist_line] 56: *** 13316
[M::ha_hist_line] 57: **** 16452
[M::ha_hist_line] 58: ***** 19650
[M::ha_hist_line] 59: ****** 24229
[M::ha_hist_line] 60: ******* 29998
[M::ha_hist_line] 61: ********* 37438
[M::ha_hist_line] 62: *********** 45813
[M::ha_hist_line] 63: ************* 54367
[M::ha_hist_line] 64: **************** 67165
[M::ha_hist_line] 65: ******************* 79086
[M::ha_hist_line] 66: *********************** 95901
[M::ha_hist_line] 67: *************************** 111990
[M::ha_hist_line] 68: ******************************* 128413
[M::ha_hist_line] 69: *********************************** 147679
[M::ha_hist_line] 70: ***************************************** 171224
[M::ha_hist_line] 71: ********************************************** 193638
[M::ha_hist_line] 72: **************************************************** 217984
[M::ha_hist_line] 73: ********************************************************** 242258
[M::ha_hist_line] 74: **************************************************************** 266643
[M::ha_hist_line] 75: ********************************************************************** 291355
[M::ha_hist_line] 76: **************************************************************************** 317522
[M::ha_hist_line] 77: ********************************************************************************* 337960
[M::ha_hist_line] 78: ************************************************************************************** 358656
[M::ha_hist_line] 79: ******************************************************************************************* 378437
[M::ha_hist_line] 80: ********************************************************************************************** 393447
[M::ha_hist_line] 81: ************************************************************************************************* 405399
[M::ha_hist_line] 82: *************************************************************************************************** 413774
[M::ha_hist_line] 83: **************************************************************************************************** 417609
[M::ha_hist_line] 84: **************************************************************************************************** 416365
[M::ha_hist_line] 85: **************************************************************************************************** 417459
[M::ha_hist_line] 86: *************************************************************************************************** 413637
[M::ha_hist_line] 87: ************************************************************************************************* 404341
[M::ha_hist_line] 88: ********************************************************************************************* 387519
[M::ha_hist_line] 89: ***************************************************************************************** 372331
[M::ha_hist_line] 90: ************************************************************************************ 352702
[M::ha_hist_line] 91: ******************************************************************************** 333308
[M::ha_hist_line] 92: ************************************************************************* 305452
[M::ha_hist_line] 93: ******************************************************************* 279706
[M::ha_hist_line] 94: ************************************************************** 257317
[M::ha_hist_line] 95: ******************************************************* 230115
[M::ha_hist_line] 96: ************************************************* 205580
[M::ha_hist_line] 97: ******************************************* 181564
[M::ha_hist_line] 98: ************************************** 159113
[M::ha_hist_line] 99: ********************************* 139005
[M::ha_hist_line] 100: ***************************** 120518
[M::ha_hist_line] 101: ************************* 102686
[M::ha_hist_line] 102: ********************* 86025
[M::ha_hist_line] 103: ****************** 73567
[M::ha_hist_line] 104: *************** 61207
[M::ha_hist_line] 105: ************ 50380
[M::ha_hist_line] 106: ********** 41491
[M::ha_hist_line] 107: ******** 34384
[M::ha_hist_line] 108: ******* 28223
[M::ha_hist_line] 109: ***** 22483
[M::ha_hist_line] 110: **** 18607
[M::ha_hist_line] 111: **** 14975
[M::ha_hist_line] 112: *** 12513
[M::ha_hist_line] 113: ** 10316
[M::ha_hist_line] 114: ** 8237
[M::ha_hist_line] 115: ** 6969
[M::ha_hist_line] 116: * 6015
[M::ha_hist_line] 117: * 5348
[M::ha_hist_line] 118: * 4850
[M::ha_hist_line] 119: * 4508
[M::ha_hist_line] 120: * 4436
[M::ha_hist_line] 121: * 4296
[M::ha_hist_line] 122: * 4655
[M::ha_hist_line] 123: * 4414
[M::ha_hist_line] 124: * 4850
[M::ha_hist_line] 125: * 5053
[M::ha_hist_line] 126: * 5326
[M::ha_hist_line] 127: * 6256
[M::ha_hist_line] 128: ** 6763
[M::ha_hist_line] 129: ** 7359
[M::ha_hist_line] 130: ** 8371
[M::ha_hist_line] 131: ** 9116
[M::ha_hist_line] 132: ** 10114
[M::ha_hist_line] 133: *** 11557
[M::ha_hist_line] 134: *** 12951
[M::ha_hist_line] 135: *** 14573
[M::ha_hist_line] 136: **** 16195
[M::ha_hist_line] 137: **** 17982
[M::ha_hist_line] 138: ***** 19859
[M::ha_hist_line] 139: ***** 22041
[M::ha_hist_line] 140: ****** 24033
[M::ha_hist_line] 141: ****** 26500
[M::ha_hist_line] 142: ******* 30035
[M::ha_hist_line] 143: ******** 32677
[M::ha_hist_line] 144: ********* 36297
[M::ha_hist_line] 145: ********* 39324
[M::ha_hist_line] 146: ********** 43146
[M::ha_hist_line] 147: *********** 47105
[M::ha_hist_line] 148: ************ 52168
[M::ha_hist_line] 149: ************* 55935
[M::ha_hist_line] 150: *************** 61001
[M::ha_hist_line] 151: **************** 65990
[M::ha_hist_line] 152: ***************** 70743
[M::ha_hist_line] 153: ****************** 74363
[M::ha_hist_line] 154: ******************* 79804
[M::ha_hist_line] 155: ******************** 83768
[M::ha_hist_line] 156: ********************* 88057
[M::ha_hist_line] 157: ********************** 92818
[M::ha_hist_line] 158: *********************** 97623
[M::ha_hist_line] 159: ************************* 103918
[M::ha_hist_line] 160: ************************** 107072
[M::ha_hist_line] 161: ************************** 110105
[M::ha_hist_line] 162: *************************** 113902
[M::ha_hist_line] 163: **************************** 117243
[M::ha_hist_line] 164: **************************** 118933
[M::ha_hist_line] 165: ***************************** 122058
[M::ha_hist_line] 166: ****************************** 123371
[M::ha_hist_line] 167: ****************************** 125091
[M::ha_hist_line] 168: ****************************** 125263
[M::ha_hist_line] 169: ****************************** 125254
[M::ha_hist_line] 170: ****************************** 123856
[M::ha_hist_line] 171: ****************************** 123656
[M::ha_hist_line] 172: ***************************** 121423
[M::ha_hist_line] 173: ***************************** 121400
[M::ha_hist_line] 174: **************************** 117980
[M::ha_hist_line] 175: **************************** 115344
[M::ha_hist_line] 176: *************************** 112655
[M::ha_hist_line] 177: ************************** 109202
[M::ha_hist_line] 178: ************************* 105297
[M::ha_hist_line] 179: ************************ 102172
[M::ha_hist_line] 180: *********************** 97507
[M::ha_hist_line] 181: ********************** 93418
[M::ha_hist_line] 182: ********************* 88400
[M::ha_hist_line] 183: ******************** 83674
[M::ha_hist_line] 184: ******************* 77971
[M::ha_hist_line] 185: ***************** 72480
[M::ha_hist_line] 186: **************** 68366
[M::ha_hist_line] 187: *************** 63165
[M::ha_hist_line] 188: ************** 58702
[M::ha_hist_line] 189: ************* 54012
[M::ha_hist_line] 190: ************ 50360
[M::ha_hist_line] 191: *********** 45887
[M::ha_hist_line] 192: ********** 40846
[M::ha_hist_line] 193: ********* 36887
[M::ha_hist_line] 194: ******** 33506
[M::ha_hist_line] 195: ******* 30266
[M::ha_hist_line] 196: ******* 27487
[M::ha_hist_line] 197: ****** 24333
[M::ha_hist_line] 198: ***** 21602
[M::ha_hist_line] 199: ***** 19303
[M::ha_hist_line] 200: **** 17108
[M::ha_hist_line] 201: **** 15156
[M::ha_hist_line] 202: *** 13661
[M::ha_hist_line] 203: *** 12076
[M::ha_hist_line] 204: *** 10526
[M::ha_hist_line] 205: ** 9215
[M::ha_hist_line] 206: ** 8143
[M::ha_hist_line] 207: ** 7395
[M::ha_hist_line] 208: ** 6602
[M::ha_hist_line] 209: * 5949
[M::ha_hist_line] 210: * 5447
[M::ha_hist_line] 211: * 4869
[M::ha_hist_line] 212: * 4270
[M::ha_hist_line] 213: * 3890
[M::ha_hist_line] 214: * 3731
[M::ha_hist_line] 215: * 3629
[M::ha_hist_line] 216: * 3494
[M::ha_hist_line] 217: * 3613
[M::ha_hist_line] 218: * 3512
[M::ha_hist_line] 219: * 3618
[M::ha_hist_line] 220: * 3772
[M::ha_hist_line] 221: * 3774
[M::ha_hist_line] 222: * 3708
[M::ha_hist_line] 223: * 3818
[M::ha_hist_line] 224: * 3986
[M::ha_hist_line] 225: * 4029
[M::ha_hist_line] 226: * 4380
[M::ha_hist_line] 227: * 4386
[M::ha_hist_line] 228: * 4510
[M::ha_hist_line] 229: * 4678
[M::ha_hist_line] 230: * 4797
[M::ha_hist_line] 231: * 5106
[M::ha_hist_line] 232: * 5197
[M::ha_hist_line] 233: * 5242
[M::ha_hist_line] 234: * 5474
[M::ha_hist_line] 235: * 5733
[M::ha_hist_line] 236: * 6021
[M::ha_hist_line] 237: * 6265
[M::ha_hist_line] 238: * 6246
[M::ha_hist_line] 239: ** 6646
[M::ha_hist_line] 240: ** 6722
[M::ha_hist_line] 241: ** 6844
[M::ha_hist_line] 242: ** 6733
[M::ha_hist_line] 243: ** 7254
[M::ha_hist_line] 244: ** 7250
[M::ha_hist_line] 245: ** 7243
[M::ha_hist_line] 246: ** 7275
[M::ha_hist_line] 247: ** 7405
[M::ha_hist_line] 248: ** 7421
[M::ha_hist_line] 249: ** 7571
[M::ha_hist_line] 250: ** 7291
[M::ha_hist_line] 251: ** 7331
[M::ha_hist_line] 252: ** 7309
[M::ha_hist_line] 253: ** 7278
[M::ha_hist_line] 254: ** 7264
[M::ha_hist_line] 255: ** 7092
[M::ha_hist_line] 256: ** 6912
[M::ha_hist_line] 257: ** 6958
[M::ha_hist_line] 258: ** 6689
[M::ha_hist_line] 259: ** 6607
[M::ha_hist_line] 260: ** 6542
[M::ha_hist_line] 261: * 6242
[M::ha_hist_line] 262: * 6185
[M::ha_hist_line] 263: * 5934
[M::ha_hist_line] 264: * 5717
[M::ha_hist_line] 265: * 5388
[M::ha_hist_line] 266: * 5448
[M::ha_hist_line] 267: * 5279
[M::ha_hist_line] 268: * 4944
[M::ha_hist_line] 269: * 4724
[M::ha_hist_line] 270: * 4549
[M::ha_hist_line] 271: * 4404
[M::ha_hist_line] 272: * 4417
[M::ha_hist_line] 273: * 4041
[M::ha_hist_line] 274: * 3888
[M::ha_hist_line] 275: * 3760
[M::ha_hist_line] 276: * 3592
[M::ha_hist_line] 277: * 3490
[M::ha_hist_line] 278: * 3150
[M::ha_hist_line] 279: * 3001
[M::ha_hist_line] 280: * 2926
[M::ha_hist_line] 281: * 3084
[M::ha_hist_line] 282: * 2758
[M::ha_hist_line] 283: * 2672
[M::ha_hist_line] 284: * 2526
[M::ha_hist_line] 285: * 2432
[M::ha_hist_line] 286: * 2313
[M::ha_hist_line] 287: * 2314
[M::ha_hist_line] 288: * 2255
[M::ha_hist_line] 289: * 2266
[M::ha_hist_line] 290: * 2230
[M::ha_hist_line] 291: * 2124
[M::ha_hist_line] 292: * 2109
[M::ha_hist_line] 293: * 2208
[M::ha_hist_line] 294: * 2187
[M::ha_hist_line] 295: * 2154
[M::ha_hist_line] 296: * 2139
[M::ha_hist_line] 297: * 2160
[M::ha_hist_line] 298: * 2211
[M::ha_hist_line] 299: * 2220
[M::ha_hist_line] 300: * 2364
[M::ha_hist_line] 301: * 2340
[M::ha_hist_line] 302: * 2367
[M::ha_hist_line] 303: * 2554
[M::ha_hist_line] 304: * 2611
[M::ha_hist_line] 305: * 2559
[M::ha_hist_line] 306: * 2525
[M::ha_hist_line] 307: * 2573
[M::ha_hist_line] 308: * 2791
[M::ha_hist_line] 309: * 2797
[M::ha_hist_line] 310: * 2819
[M::ha_hist_line] 311: * 2945
[M::ha_hist_line] 312: * 2958
[M::ha_hist_line] 313: * 3034
[M::ha_hist_line] 314: * 3275
[M::ha_hist_line] 315: * 3267
[M::ha_hist_line] 316: * 3351
[M::ha_hist_line] 317: * 3284
[M::ha_hist_line] 318: * 3513
[M::ha_hist_line] 319: * 3510
[M::ha_hist_line] 320: * 3692
[M::ha_hist_line] 321: * 3704
[M::ha_hist_line] 322: * 3755
[M::ha_hist_line] 323: * 3906
[M::ha_hist_line] 324: * 3910
[M::ha_hist_line] 325: * 3888
[M::ha_hist_line] 326: * 4038
[M::ha_hist_line] 327: * 4052
[M::ha_hist_line] 328: * 4249
[M::ha_hist_line] 329: * 4048
[M::ha_hist_line] 330: * 3908
[M::ha_hist_line] 331: * 4098
[M::ha_hist_line] 332: * 3993
[M::ha_hist_line] 333: * 4066
[M::ha_hist_line] 334: * 4055
[M::ha_hist_line] 335: * 4079
[M::ha_hist_line] 336: * 4027
[M::ha_hist_line] 337: * 3871
[M::ha_hist_line] 338: * 3942
[M::ha_hist_line] 339: * 3919
[M::ha_hist_line] 340: * 3896
[M::ha_hist_line] 341: * 3891
[M::ha_hist_line] 342: * 3721
[M::ha_hist_line] 343: * 3773
[M::ha_hist_line] 344: * 3657
[M::ha_hist_line] 345: * 3596
[M::ha_hist_line] 346: * 3377
[M::ha_hist_line] 347: * 3273
[M::ha_hist_line] 348: * 3190
[M::ha_hist_line] 349: * 3224
[M::ha_hist_line] 350: * 3142
[M::ha_hist_line] 351: * 3076
[M::ha_hist_line] 352: * 3058
[M::ha_hist_line] 353: * 2916
[M::ha_hist_line] 354: * 2776
[M::ha_hist_line] 355: * 2764
[M::ha_hist_line] 356: * 2785
[M::ha_hist_line] 357: * 2701
[M::ha_hist_line] 358: * 2490
[M::ha_hist_line] 359: * 2416
[M::ha_hist_line] 360: * 2361
[M::ha_hist_line] 361: * 2298
[M::ha_hist_line] 362: * 2325
[M::ha_hist_line] 363: * 2146
[M::ha_hist_line] 364: * 2136
[M::ha_hist_line] 365: * 2099
[M::ha_hist_line] rest: *********************************************************************************************** 396842
[M::ha_analyze_count] left: none
[M::ha_analyze_count] right: none
[M::ha_pt_gen] peak_hom: 83; peak_het: -1
[M::ha_ct_shrink::285039.075*35.33] ==> counted 17036513 distinct minimizer k-mers
[M::ha_pt_gen::] counting in normal mode
[M::yak_count] collected 2137320573 minimizers
[M::ha_pt_gen::285482.798*35.31] ==> indexed 2135588024 positions, counted 17036513 distinct minimizer k-mers
[M::ha_assemble::297514.365*35.34@246.283GB] ==> found overlaps for the final round
[M::ha_print_ovlp_stat] # overlaps: 1183659490
[M::ha_print_ovlp_stat] # strong overlaps: 596745657
[M::ha_print_ovlp_stat] # weak overlaps: 586913833
[M::ha_print_ovlp_stat] # exact overlaps: 1149035029
[M::ha_print_ovlp_stat] # inexact overlaps: 34624461
[M::ha_print_ovlp_stat] # overlaps without large indels: 1180991551
[M::ha_print_ovlp_stat] # reverse overlaps: 410771757
Writing reads to disk...
Reads has been written.
Thats a lot of information. The Hifiasm output page says that for heterozygous samples (which mine likely are), there should be 2 peaks in the k-mer plot, where the smaller peak is around the heterozygous read coverage and the larger peak is around the homozygous read coverage. This is true for all k-mer plots produced from this data. In all of my k-mer plots, the homozygous peak is 83 and the heterozygous peak is 168. I’m going to include the information without the k-mer plots below because they are so large:
[M::ha_analyze_count] lowest: count[17] = 11309
[M::ha_analyze_count] highest: count[85] = 9499858
## first k-mer plot
[M::ha_analyze_count] left: none
[M::ha_analyze_count] right: count[168] = 3093394
[M::ha_ft_gen] peak_hom: 168; peak_het: 85
[M::ha_ct_shrink::3427.856*4.32] ==> counted 2684382 distinct minimizer k-mers
[M::ha_ft_gen::3431.212*4.31@20.882GB] ==> filtered out 2684382 k-mers occurring 840 or more times
[M::ha_opt_update_cov] updated max_n_chain to 840
[M::yak_count] collected 2139678804 minimizers
[M::ha_pt_gen::4355.642*5.80] ==> counted 31723566 distinct minimizer k-mers
[M::ha_pt_gen] count[4095] = 0 (for sanity check)
[M::ha_analyze_count] lowest: count[17] = 2230
[M::ha_analyze_count] highest: count[83] = 418590
## second k-mer plot
[M::ha_analyze_count] left: none
[M::ha_analyze_count] right: count[168] = 125423
[M::ha_pt_gen] peak_hom: 168; peak_het: 83
[M::ha_ct_shrink::4355.907*5.81] ==> counted 17720475 distinct minimizer k-mers
[M::ha_pt_gen::] counting in normal mode
[M::yak_count] collected 2139678804 minimizers
[M::ha_pt_gen::4818.141*7.37] ==> indexed 2125675713 positions, counted 17720475 distinct minimizer k-mers
[M::ha_assemble::100006.981*34.52@107.237GB] ==> corrected reads for round 1
[M::ha_assemble] # bases: 79183709778; # corrected bases: 91988177; # recorrected bases: 115414
[M::ha_assemble] size of buffer: 61.709GB
[M::yak_count] collected 2137476150 minimizers
[M::ha_pt_gen::100401.244*34.48] ==> counted 19037497 distinct minimizer k-mers
[M::ha_pt_gen] count[4095] = 0 (for sanity check)
[M::ha_analyze_count] lowest: count[13] = 1719
[M::ha_analyze_count] highest: count[83] = 417869
## third k-mer plot
[M::ha_analyze_count] left: none
[M::ha_analyze_count] right: none
[M::ha_pt_gen] peak_hom: 83; peak_het: -1
[M::ha_ct_shrink::100401.532*34.48] ==> counted 17060420 distinct minimizer k-mers
[M::ha_pt_gen::] counting in normal mode
[M::yak_count] collected 2137476150 minimizers
[M::ha_pt_gen::100851.319*34.43] ==> indexed 2135499073 positions, counted 17060420 distinct minimizer k-mers
[M::ha_assemble::192251.063*35.13@143.827GB] ==> corrected reads for round 2
[M::ha_assemble] # bases: 79186546424; # corrected bases: 1694280; # recorrected bases: 16078
[M::ha_assemble] size of buffer: 60.552GB
[M::yak_count] collected 2137334800 minimizers
[M::ha_pt_gen::192627.741*35.11] ==> counted 18787923 distinct minimizer k-mers
[M::ha_pt_gen] count[4095] = 0 (for sanity check)
[M::ha_analyze_count] lowest: count[9] = 2352
[M::ha_analyze_count] highest: count[83] = 417664
## fourth k-mer plot
[M::ha_analyze_count] left: none
[M::ha_analyze_count] right: none
[M::ha_pt_gen] peak_hom: 83; peak_het: -1
[M::ha_ct_shrink::192628.074*35.11] ==> counted 17040202 distinct minimizer k-mers
[M::ha_pt_gen::] counting in normal mode
[M::yak_count] collected 2137334800 minimizers
[M::ha_pt_gen::193066.706*35.08] ==> indexed 2135587079 positions, counted 17040202 distinct minimizer k-mers
[M::ha_assemble::284661.787*35.35@241.898GB] ==> corrected reads for round 3
[M::ha_assemble] # bases: 79186730278; # corrected bases: 235080; # recorrected bases: 12650
[M::ha_assemble] size of buffer: 60.546GB
[M::yak_count] collected 2137320573 minimizers
[M::ha_pt_gen::285038.830*35.33] ==> counted 18769062 distinct minimizer k-mers
[M::ha_pt_gen] count[4095] = 0 (for sanity check)
[M::ha_analyze_count] lowest: count[9] = 2315
[M::ha_analyze_count] highest: count[83] = 417609
## fifth k-mer plot
[M::ha_analyze_count] left: none
[M::ha_analyze_count] right: none
[M::ha_pt_gen] peak_hom: 83; peak_het: -1
[M::ha_ct_shrink::285039.075*35.33] ==> counted 17036513 distinct minimizer k-mers
[M::ha_pt_gen::] counting in normal mode
[M::yak_count] collected 2137320573 minimizers
[M::ha_pt_gen::285482.798*35.31] ==> indexed 2135588024 positions, counted 17036513 distinct minimizer k-mers
[M::ha_assemble::297514.365*35.34@246.283GB] ==> found overlaps for the final round
[M::ha_print_ovlp_stat] # overlaps: 1183659490
[M::ha_print_ovlp_stat] # strong overlaps: 596745657
[M::ha_print_ovlp_stat] # weak overlaps: 586913833
[M::ha_print_ovlp_stat] # exact overlaps: 1149035029
[M::ha_print_ovlp_stat] # inexact overlaps: 34624461
[M::ha_print_ovlp_stat] # overlaps without large indels: 1180991551
[M::ha_print_ovlp_stat] # reverse overlaps: 410771757
Writing reads to disk...
Reads has been written.
Writing ma_hit_ts to disk...
ma_hit_ts has been written.
Writing ma_hit_ts to disk...
ma_hit_ts has been written.
bin files have been written.
[M::purge_dups] homozygous read coverage threshold: 168
[M::purge_dups] purge duplication coverage threshold: 210
Writing raw unitig GFA to disk...
Writing processed unitig GFA to disk...
[M::purge_dups] homozygous read coverage threshold: 168
[M::purge_dups] purge duplication coverage threshold: 210
[M::mc_solve_core::0.284] ==> Partition
[M::adjust_utg_by_primary] primary contig coverage range: [142, infinity]
Writing apul.hifiasm.bp.p_ctg.gfa to disk...
[M::adjust_utg_by_trio] primary contig coverage range: [142, infinity]
Writing apul.hifiasm.bp.hap1.p_ctg.gfa to disk...
[M::adjust_utg_by_trio] primary contig coverage range: [142, infinity]
Writing apul.hifiasm.bp.hap2.p_ctg.gfa to disk...
Inconsistency threshold for low-quality regions in BED files: 70%
[M::main] Version: 0.16.1-r375
[M::main] CMD: hifiasm -o apul.hifiasm -t 36 m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq
[M::main] Real time: 304623.035 sec; CPU: 10520661.634 sec; Peak RSS: 246.283 GB
Okay so 4 rounds of assembly were done, hypothetically improving the assembly each time. Weirdly, the first 2 rounds had the heterozygous peak at 68, but the last couple of rounds had the heterozygous peak at -1. In all k-mer plots, there is a high peak at the 1 location, but this doesn’t seem unusual (based on the example posts here and here provided by the hifiasm log interpretation section). Overall, I think this file is providing information on the assembly iterations.
Given all of this information, let’s now run BUSCO to assess completeness of assembly. In the scripts folder: nano busco_unfilt_hifiasm.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.bp.p_ctg.gfa > apul.hifiasm.bp.p_ctg.fa
echo "Begin busco on unfiltered hifiasm-assembled fasta" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.bp.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.busco.canu -m genome
echo "busco complete for unfiltered hifiasm-assembled fasta" $(date)
Submitted batch job 305426. I could also run some analysis on the hap assemblies, but I’m going to wait until I am done with the filtering and re-assembly, as that assembly is the one that I will likely be moving forward with. How are they making the distinction between hap1/father and hap2/mother?
Busco ran in ~45 mins and results look a lot better than they did with canu! This is the primary result from the output file:
2024-03-11 13:37:21 INFO:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.3%[S:92.0%,D:1.3%],F:3.1%,M:3.6%,n:954 |
|890 Complete BUSCOs (C) |
|878 Complete and single-copy BUSCOs (S) |
|12 Complete and duplicated BUSCOs (D) |
|30 Fragmented BUSCOs (F) |
|34 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
This looks so much better than the canu assembly! The previous canu assembly was 94.4% complete, but had only 9.4% single copy BUSCOs and 85% duplicated BUSCOs. Ideally, the duplication level should be lower. The hifiasm assembly had 93.3% completeness, 92% single copy BUSCOs, and 1.3% duplicated BUSCOs. Hifiasm is definitely the way to go for assembly. Now I just have to wait until the blast prok is done so I can remove any contamination. After I remove contamination, I will re-assemble using hifiasm.
20240313
Blast prok failed after 2 days and then restarted on Andromeda. Maybe I should stop the code after 2 days…idk. Maybe I need to increase the memory for the job? Canceling the job (305351) and increasing the memory (#SBATCH --mem=500GB). Submitted batch job 308997
20240316
Copied the prok data to my local computer. It is still running on the server, but I’m nervous it will restart again. If it does restart, I’ll cancel the job and just use this data from today. On my local computer, I combined the prok and viral blast results and then removed any hits whose bit score was <1000.
cd /Users/jillashey/Desktop/PutnamLab/Apulchra_genome
cat viral_contaminant_hits_rr.txt prok_contaminant_hits_rr.txt > all_contaminant_hits_rr.txt
awk '$12 > 1000 {print $0}' all_contaminant_hits_rr.txt > contaminant_hits_pv_passfilter_rr.txt
I then looked at the contamination hits in R. See code here.
As a summary, I first read in the eukaryotic blast hits that passed the contamination threshold. I found that only 2 reads had any euk contamination (m84100_240128_024355_s2/48759857/ccs and m84100_240128_024355_s2/234751852/ccs). I then read in the prokaryotic and viral blast hits that passed the threshold (only 224 blast hits passed). I calculated the percentage of each hits align length to the contigs so if there was a result that had 100%, that would mean that the whole contig was a contaminant. I looked at a histogram of the % alignments and found that most of the % alignments are on the lower size and there are not many 100% sequences. I summarized the contigs that were to be filtered out and found that 222 contigs had some level of pv contamination. I added the euk + pv contamination reads together (224 total) and calculated the proportion of contamination to raw reads. The contamination ended up being only 0.003797649% of the raw reads, which is pretty amazing! I calculated the mean length of the filtered reads (13,242.1 bp) and the sums of the unfiltered read length (79183709778 total bp) and filtered read length (79181142809 total bp). Using these lengths, I calculated a rough estimation of sequencing depth and found we have roughly 100x coverage! That’s similar to the Young et al. 2024 results as well. Finally, I wrote the list of filtered reads to a text file on my local computer. This information will be used to filter the raw reads on Andromeda prior to assembly. I’m impressed with the low contamination and the high coverage of the PacBio HiFi reads.
20240319
Prok blast script finally finished running! Took about 5 days. Cat the prok and viral results together and remove anything that has a bit score <1000.
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
cat viral_contaminant_hits_rr.txt prok_contaminant_hits_rr.txt > all_contaminant_hits_rr.txt
awk '$12 > 1000 {print $0}' all_contaminant_hits_rr.txt > contaminant_hits_pv_passfilter_rr.txt
Similarly to what I did above, I looked at the contamination hits in R. See code here.
As a summary, I first read in the eukaryotic blast hits that passed the contamination threshold. I found that only 2 reads had any euk contamination (m84100_240128_024355_s2/48759857/ccs and m84100_240128_024355_s2/234751852/ccs). I then read in the prokaryotic and viral blast hits that passed the threshold (only 2 blast hits passed). I calculated the percentage of each hits align length to the contigs so if there was a result that had 100%, that would mean that the whole contig was a contaminant. I looked at a histogram of the % alignments and found that most of the % alignments are on the lower size and there are not many 100% sequences. I summarized the contigs that were to be filtered out and found that 494 contigs had some level of pv contamination. I added the euk + pv contamination reads together (496 contigs total) and calculated the proportion of contamination to raw reads. The contamination ended up being only 0.00840908% of the raw reads, which is pretty amazing! I calculated the mean length of the filtered reads (13,424.84 bp) and the sums of the unfiltered read length (79183709778 total bp) and filtered read length (79178244314 total bp). Using these lengths, I calculated a rough estimation of sequencing depth and found we have roughly 100x coverage! That’s similar to the Young et al. 2024 results as well. Finally, I wrote the list of filtered reads to a text file on my local computer. This information will be used to filter the raw reads on Andromeda prior to assembly. I’m impressed with the low contamination and the high coverage of the PacBio HiFi reads.
20240320
Before filtered the hifi reads, I’m going to clean up the /data/putnamlab/jillashey/Apul_Genome/assembly/data folder so that I have more memory for the next steps. Here’s whats in there right now:
total 312G
-rw-r--r--. 1 jillashey 148G Feb 8 02:37 m84100_240128_024355_s2.hifi_reads.bc1029.fastq.fastq
-rwxr-xr-x. 1 jillashey 1.1K Feb 8 14:13 apul.seqStore.sh
-rw-r--r--. 1 jillashey 951 Feb 8 14:31 apul.seqStore.err
drwxr-xr-x. 3 jillashey 4.0K Feb 8 14:32 apul.seqStore
-rw-r--r--. 1 jillashey 23K Feb 9 01:47 apul.report
-rw-r--r--. 1 jillashey 7.0M Feb 9 01:51 apul.contigs.layout.tigInfo
-rw-r--r--. 1 jillashey 155M Feb 9 01:51 apul.contigs.layout.readToTig
-rw-r--r--. 1 jillashey 2.8G Feb 9 01:57 apul.unassembled.fasta
-rw-r--r--. 1 jillashey 943M Feb 9 02:01 apul.contigs.fasta
drwxr-xr-x. 2 jillashey 4.0K Feb 13 14:01 busco_output
drwxr-xr-x. 3 jillashey 4.0K Feb 13 14:01 busco_downloads
-rw-r--r--. 1 jillashey 7.2K Feb 13 14:02 busco_96228.log
lrwxrwxrwx. 1 jillashey 108 Feb 20 14:08 m84100_240128_024355_s2.hifi_reads.bc1029.bam -> /data/putnamlab/KITT/hputnam/20240129_Apulchra_Genome_LongRead/m84100_240128_024355_s2.hifi_reads.bc1029.bam
-rw-r--r--. 1 jillashey 106 Feb 21 16:48 pb.fofn
drwxr-xr-x. 9 jillashey 4.0K Feb 21 16:53 unitigging
-rw-r--r--. 1 jillashey 74G Mar 1 16:08 m84100_240128_024355_s2.hifi_reads.bc1029.fasta
-rw-r--r--. 1 jillashey 244M Mar 1 16:36 rr_read_lengths.txt
-rw-r--r--. 1 jillashey 106K Mar 3 16:27 contaminant_hits_euks_rr.txt
-rw-r--r--. 1 jillashey 2.4K Mar 3 16:29 contaminants_pass_filter_euks_rr.txt
-rw-r--r--. 1 jillashey 69M Mar 5 23:20 viral_contaminant_hits_rr.txt
-rw-r--r--. 1 jillashey 19G Mar 7 23:58 apul.hifiasm.ec.bin
-rw-r--r--. 1 jillashey 47G Mar 8 00:08 apul.hifiasm.ovlp.source.bin
-rw-r--r--. 1 jillashey 17G Mar 8 00:12 apul.hifiasm.ovlp.reverse.bin
-rw-r--r--. 1 jillashey 1.2G Mar 8 01:37 apul.hifiasm.bp.r_utg.gfa
-rw-r--r--. 1 jillashey 21M Mar 8 01:37 apul.hifiasm.bp.r_utg.noseq.gfa
-rw-r--r--. 1 jillashey 8.6M Mar 8 01:41 apul.hifiasm.bp.r_utg.lowQ.bed
-rw-r--r--. 1 jillashey 1.1G Mar 8 01:42 apul.hifiasm.bp.p_utg.gfa
-rw-r--r--. 1 jillashey 21M Mar 8 01:42 apul.hifiasm.bp.p_utg.noseq.gfa
-rw-r--r--. 1 jillashey 8.2M Mar 8 01:46 apul.hifiasm.bp.p_utg.lowQ.bed
-rw-r--r--. 1 jillashey 506M Mar 8 01:47 apul.hifiasm.bp.p_ctg.gfa
-rw-r--r--. 1 jillashey 11M Mar 8 01:47 apul.hifiasm.bp.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 2.0M Mar 8 01:49 apul.hifiasm.bp.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 469M Mar 8 01:50 apul.hifiasm.bp.hap1.p_ctg.gfa
-rw-r--r--. 1 jillashey 9.9M Mar 8 01:50 apul.hifiasm.bp.hap1.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 2.0M Mar 8 01:52 apul.hifiasm.bp.hap1.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 468M Mar 8 01:52 apul.hifiasm.bp.hap2.p_ctg.gfa
-rw-r--r--. 1 jillashey 9.9M Mar 8 01:52 apul.hifiasm.bp.hap2.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 1.9M Mar 8 01:54 apul.hifiasm.bp.hap2.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 495M Mar 11 12:52 apul.hifiasm.bp.p_ctg.fa
-rw-r--r--. 1 jillashey 246M Mar 19 14:05 prok_contaminant_hits_rr.txt
-rw-r--r--. 1 jillashey 315M Mar 19 16:08 all_contaminant_hits_rr.txt
-rw-r--r--. 1 jillashey 1.1M Mar 19 16:09 contaminant_hits_pv_passfilter_rr.txt
I removed the following:
rm -r apul.seqStore
rm apul*
rm -r busco*
rm pb.fofn
rm -r unitigging/
Now I have more space. Copy the file all_contam_rem_good_hifi_read_list.txt that was generated from the R code mentioned above. This specific file was written starting on line 242. It contains the reads that have passed contamination filtering. I copied this file into /data/putnamlab/jillashey/Apul_Genome/assembly/data.
wc -l all_contam_rem_good_hifi_read_list.txt
5897892 all_contam_rem_good_hifi_read_list.txt
The vast majority of the hifi reads are retained after contamination filtering, which is a good sign of high quality sequencing. My next step is to subset the raw hifi fasta file to remove the contaminants identified above. I can do this with the seqtk subseq command. In the scripts folder: nano subseq.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load seqtk/1.3-GCC-9.3.0
echo "Subsetting hifi reads that passed contamination filtering" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
seqtk subseq m84100_240128_024355_s2.hifi_reads.bc1029.fasta all_contam_rem_good_hifi_read_list.txt > hifi_rr_allcontam_rem.fasta
echo "Subsetting complete!" $(date)
Submitted batch job 309659. Finished in about 8 mins but the output file looks like this:
>m84100_240128_024355_s2/261887593/ccs:18224-18224
A
>m84100_240128_024355_s2/255530003/ccs:21870-21870
A
>m84100_240128_024355_s2/249237028/ccs:23691-23691
A
>m84100_240128_024355_s2/262606536/ccs:14772-14772
A
>m84100_240128_024355_s2/217322854/ccs:12923-12923
A
>m84100_240128_024355_s2/256512826/ccs:12914-12914
A
>m84100_240128_024355_s2/245632166/ccs:28440-28440
A
>m84100_240128_024355_s2/250548903/ccs:23076-23076
A
>m84100_240128_024355_s2/256054930/ccs:15405-15405
A
>m84100_240128_024355_s2/241242930/ccs:15521-15521
A
>m84100_240128_024355_s2/254348773/ccs:14578-14578
C
>m84100_240128_024355_s2/252319399/ccs:12407-12407
A
>m84100_240128_024355_s2/229183717/ccs:5757-5757
Not ideal. It looks like it only took the first letter from each sequence. Maybe I need to remove the length information from the all_contam_rem_good_hifi_read_list.txt file?
awk '{$2=""; print $0}' all_contam_rem_good_hifi_read_list.txt > output_file.txt
Edit the script so that the list of reads to keep is output_file.txt and decrease mem to 250GB. Submitted batch job 309672. This appears to have worked!
zgrep -c ">" hifi_rr_allcontam_rem.fasta
5897892
Following Young et al. 2024, I need to use Merqury and GenomeScope2 analysis of the cleaned reads. Merqury and Meryl seem to be related somehow, but not sure. Meryl is a tool for counting and working with sets of k-mers. It is a part of Canu as well. So it seems like Meryl counts the k-mers and Merqury estimates accuracy and completeness? Young et al. 2024 used only meryl here to generate a kmer database, which was then used as input to genomescope2. Merqury was used after hifiasm assembly.
First, the best value for k needs to be determined for use in Meryl. This can be done with the best_k.sh script from Merqury. I’m going to copy this script into my own scripts folder. Young et al. 2024 used 500mb estimated genome size. This is what I will use too, as previous coral/Acropora genomes are about this size. I will need to install Meryl and Merqury first. I’m following the Meryl and Merqury githubs for installation instructions.
For Meryl:
wget https://github.com/marbl/meryl/releases/download/v1.4.1/meryl-1.4.1.Linux-amd64.tar.xz
tar -xJf meryl-1.4.1.Linux-amd64.tar.xz
export PATH=/data/putnamlab/jillashey/Apul_Genome/assembly/meryl-1.4.1/bin:$PATH
Now that I have exported the PATH variable to include the directory where the Meryl executable files are, I can run Meryl commands (I think), regardless of working directory. For now, lets run meryl with k=18. The k-mer database will be generated using k=18 and the cleaned reads. In the scripts folder: nano meryl.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=1
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
export PATH=/data/putnamlab/jillashey/Apul_Genome/assembly/meryl-1.4.1/bin:$PATH
echo "Creating meryl k-mer db" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
meryl k=18 \
count hifi_rr_allcontam_rem.fasta \
output meryl_merc
echo "Meryl k-mer db complete!" $(date)
Submitted batch job 309682. While this runs, lets try to install Merqury.
git clone https://github.com/marbl/merqury.git
cd merqury
export MERQURY=$PWD:$PATH
Hypothetically, Merqury is now installed. Perhaps now we can run the best k script? I am not sure what genome size estimate to use. Shinzato et al. 2020 assessed several Acropora genomes and found the genome size ranged from 384 (Acropora microphthalma) to 447 (Acropora hyacinthus) Mb. I think I will go with 450 Mb. Let’s try to run the best k script.
$MERQURY/best_k.sh 450000000
Giving me this error:
-bash: /data/putnamlab/jillashey/Apul_Genome/assembly/merqury:/data/putnamlab/jillashey/Apul_Genome/assembly/meryl-1.4.1/bin:/path/to/meryl-1.4.1/bin:/path/to/meryl/…/bin:/opt/software/Miniconda3/4.9.2/bin:/opt/software/Miniconda3/4.9.2/condabin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home/jillashey/.local/bin:/home/jillashey/bin/best_k.sh: No such file or directory
I’m not sure what it means or how to fix it. On the Merqury github, it lists dependencies that Merqury may need to run:
- gcc 10.2.0 or higher (for installing Meryl)
- Meryl v1.4.1
- Java run time environment (JRE)
- R with argparse, ggplot2, and scales (recommend R 4.0.3+)
- bedtools
- samtools
Maybe I need to load these? Idk I am confused. I may just continue with the initial assembly…because I don’t really see the importance of this step. In Young et al. 2024, it says “the kmer profile of cleaned raw HiFi reads was generated with Meryl [34], and used for genome profiling with GenomeScope2 [69] to estimate genome size, repetitiveness, heterozygosity, and ploidy.” I will come back to this. I may need to email Kevin Bryan to install meryl, merqury and genomescope2.
While I wait for his response, I will start running the initial hifiasm assembly with default flags. I have a script for hifiasm on the server, but I’m going to make a new one for the initial assembly. In the scripts folder: nano initial_hifiasm.sh
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/hifiasm
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting assembly with hifiasm" $(date)
hifiasm -o apul.hifiasm.intial hifi_rr_allcontam_rem.fasta -t 36 2> apul_hifiasm_allcontam_rem_initial.asm.log
echo "Assembly with hifiasm complete!" $(date)
conda deactivate
Submitted batch job 309689
20240325
Initial assembly ran in about 3 days. These are the files that were generated:
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
-rw-r--r--. 1 jillashey 19G Mar 24 02:10 apul.hifiasm.intial.ec.bin
-rw-r--r--. 1 jillashey 47G Mar 24 02:20 apul.hifiasm.intial.ovlp.source.bin
-rw-r--r--. 1 jillashey 17G Mar 24 02:23 apul.hifiasm.intial.ovlp.reverse.bin
-rw-r--r--. 1 jillashey 1.2G Mar 24 03:40 apul.hifiasm.intial.bp.r_utg.gfa
-rw-r--r--. 1 jillashey 21M Mar 24 03:40 apul.hifiasm.intial.bp.r_utg.noseq.gfa
-rw-r--r--. 1 jillashey 8.6M Mar 24 03:44 apul.hifiasm.intial.bp.r_utg.lowQ.bed
-rw-r--r--. 1 jillashey 1.1G Mar 24 03:45 apul.hifiasm.intial.bp.p_utg.gfa
-rw-r--r--. 1 jillashey 21M Mar 24 03:45 apul.hifiasm.intial.bp.p_utg.noseq.gfa
-rw-r--r--. 1 jillashey 8.2M Mar 24 03:49 apul.hifiasm.intial.bp.p_utg.lowQ.bed
-rw-r--r--. 1 jillashey 506M Mar 24 03:50 apul.hifiasm.intial.bp.p_ctg.gfa
-rw-r--r--. 1 jillashey 11M Mar 24 03:50 apul.hifiasm.intial.bp.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 2.0M Mar 24 03:52 apul.hifiasm.intial.bp.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 469M Mar 24 03:52 apul.hifiasm.intial.bp.hap1.p_ctg.gfa
-rw-r--r--. 1 jillashey 9.9M Mar 24 03:52 apul.hifiasm.intial.bp.hap1.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 2.0M Mar 24 03:54 apul.hifiasm.intial.bp.hap1.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 468M Mar 24 03:55 apul.hifiasm.intial.bp.hap2.p_ctg.gfa
-rw-r--r--. 1 jillashey 9.9M Mar 24 03:55 apul.hifiasm.intial.bp.hap2.p_ctg.noseq.gfa
-rw-r--r--. 1 jillashey 1.9M Mar 24 03:56 apul.hifiasm.intial.bp.hap2.p_ctg.lowQ.bed
-rw-r--r--. 1 jillashey 80K Mar 24 03:57 apul_hifiasm_allcontam_rem_initial.asm.log
Many files. The output file description can be found above and also on the hifiasm output website. The log output file contains the k-mer histogram (similar to what is posted above), which shows two peaks, indicative of a heterozygous genome assembly. This is what the bottom of the log file looks like:
[M::ha_pt_gen::] counting in normal mode
[M::yak_count] collected 2137171007 minimizers
[M::ha_pt_gen::281249.073*35.34] ==> indexed 2135566893 positions, counted 17030417 distinct minimizer k-mers
[M::ha_assemble::292970.825*35.36@202.994GB] ==> found overlaps for the final round
[M::ha_print_ovlp_stat] # overlaps: 1183659340
[M::ha_print_ovlp_stat] # strong overlaps: 596745652
[M::ha_print_ovlp_stat] # weak overlaps: 586913688
[M::ha_print_ovlp_stat] # exact overlaps: 1149035007
[M::ha_print_ovlp_stat] # inexact overlaps: 34624333
[M::ha_print_ovlp_stat] # overlaps without large indels: 1180991403
[M::ha_print_ovlp_stat] # reverse overlaps: 410771728
Writing reads to disk...
Reads has been written.
Writing ma_hit_ts to disk...
ma_hit_ts has been written.
Writing ma_hit_ts to disk...
ma_hit_ts has been written.
bin files have been written.
[M::purge_dups] homozygous read coverage threshold: 168
[M::purge_dups] purge duplication coverage threshold: 210
Writing raw unitig GFA to disk...
Writing processed unitig GFA to disk...
[M::purge_dups] homozygous read coverage threshold: 168
[M::purge_dups] purge duplication coverage threshold: 210
[M::mc_solve_core::0.308] ==> Partition
[M::adjust_utg_by_primary] primary contig coverage range: [142, infinity]
Writing apul.hifiasm.intial.bp.p_ctg.gfa to disk...
[M::adjust_utg_by_trio] primary contig coverage range: [142, infinity]
[M::adjust_utg_by_trio] primary contig coverage range: [142, infinity]
Writing apul.hifiasm.intial.bp.hap1.p_ctg.gfa to disk...
[M::adjust_utg_by_trio] primary contig coverage range: [142, infinity]
Writing apul.hifiasm.intial.bp.hap2.p_ctg.gfa to disk...
Inconsistency threshold for low-quality regions in BED files: 70%
[M::main] Version: 0.16.1-r375
[M::main] CMD: hifiasm -o apul.hifiasm.intial -t 36 hifi_rr_allcontam_rem.fasta
[M::main] Real time: 299500.413 sec; CPU: 10366612.139 sec; Peak RSS: 202.994 GB
Does the M::purge_dups mean that this is the value that I should be using for the purge_dups flag in hifiasm? It gives me two lines for M::purge_dups: homozygous read coverage threshold: 168 and purge duplication coverage threshold: 210. I may need to talk with Ross and Hollie more about this. I’m going to QC the assembly and the haplotype assemblies with busco and quast. In the scripts folder: nano initial_qc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.intial.bp.p_ctg.gfa > apul.hifiasm.intial.bp.p_ctg.fa
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.intial.bp.hap1.p_ctg.gfa > apul.hifiasm.intial.bp.hap1.p_ctg.fa
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.intial.bp.hap2.p_ctg.gfa > apul.hifiasm.intial.bp.hap2.p_ctg.fa
echo "Begin busco on filtered hifiasm-assembled fasta (initial run)" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.intial.bp.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.initial.busco -m genome
echo "busco complete for unfiltered hifiasm-assembled fasta (initial run)" $(date)
echo "Begin busco on hifiasm-assembled haplotype 1 fasta" $(date)
# Reset query
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.intial.bp.hap1.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.initial.hap1.busco -m genome
echo "busco complete for hifiasm-assembled haplotype 1 fasta (initial run)" $(date)
echo "Begin busco on hifiasm-assembled haplotype 2 fasta" $(date)
# Reset query
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.intial.bp.hap2.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.initial.hap2.busco -m genome
echo "busco complete for hifiasm-assembled haplotype 2 fasta (initial run)" $(date)
echo "busco complete all assemblies of interest (initial run)" $(date)
echo "Begin quast of primary and haplotypes (initial run)" $(date)
module load QUAST/5.2.0-foss-2021b
# there is another version of quast if the one above does not work: QUAST/5.0.2-foss-2020b-Python-2.7.18
quast -t 15 --eukaryote \
apul.hifiasm.intial.bp.p_ctg.fa \
apul.hifiasm.intial.bp.hap1.p_ctg.fa \
apul.hifiasm.intial.bp.hap2.p_ctg.fa \
/data/putnamlab/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta \
-o /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast
echo "Quast complete (initial run); all QC complete!" $(date)
Submitted batch job 309969. Ran in ~2 hours. Busco ran but only for the primariy assembly. For some reason, the hap1 and hap2 busco did not run. Quast appears to have failed with these errors:
foss/2021b(24):ERROR:150: Module 'foss/2021b' conflicts with the currently loaded module(s) 'foss/2020b'
foss/2021b(24):ERROR:102: Tcl command execution failed: conflict foss
Python/3.9.6-GCCcore-11.2.0(61):ERROR:150: Module 'Python/3.9.6-GCCcore-11.2.0' conflicts with the currently loaded module(s) 'Python/3.8.6-GCCcore-10.2.0'
Python/3.9.6-GCCcore-11.2.0(61):ERROR:102: Tcl command execution failed: conflict Python
Perl/5.34.0-GCCcore-11.2.0(133):ERROR:150: Module 'Perl/5.34.0-GCCcore-11.2.0' conflicts with the currently loaded module(s) 'Perl/5.32.0-GCCcore-10.2.0'
Perl/5.34.0-GCCcore-11.2.0(133):ERROR:102: Tcl command execution failed: conflict Perl
foss/2021b(24):ERROR:150: Module 'foss/2021b' conflicts with the currently loaded module(s) 'foss/2020b'
foss/2021b(24):ERROR:102: Tcl command execution failed: conflict foss
Python/3.9.6-GCCcore-11.2.0(61):ERROR:150: Module 'Python/3.9.6-GCCcore-11.2.0' conflicts with the currently loaded module(s) 'Python/3.8.6-GCCcore-10.2.0'
Python/3.9.6-GCCcore-11.2.0(61):ERROR:102: Tcl command execution failed: conflict Python
SciPy-bundle/2021.10-foss-2021b(30):ERROR:150: Module 'SciPy-bundle/2021.10-foss-2021b' conflicts with the currently loaded module(s) 'SciPy-bundle/2020.11-foss-2020b'
SciPy-bundle/2021.10-foss-2021b(30):ERROR:102: Tcl command execution failed: conflict SciPy-bundle
GCCcore/11.2.0(24):ERROR:150: Module 'GCCcore/11.2.0' conflicts with the currently loaded module(s) 'GCCcore/10.2.0'
GCCcore/11.2.0(24):ERROR:102: Tcl command execution failed: conflict GCCcore
Basically just a lot of conflicting modules. Below are the results for the Busco code for the primary assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.3%[S:92.0%,D:1.3%],F:3.1%,M:3.6%,n:954 |
|890 Complete BUSCOs (C) |
|878 Complete and single-copy BUSCOs (S) |
|12 Complete and duplicated BUSCOs (D) |
|30 Fragmented BUSCOs (F) |
|34 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Going to edit the initial_qc.sh script so that I am including all things for busco to run properly. I’m also commenting out the lines that ran successfully and switching the module to QUAST/5.0.2-foss-2020b-Python-2.7.18. Submitted batch job 309984. It ran, but only the hap1 busco scores were generated, not hap2. Below are the results for the hap1 assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.3%[S:92.8%,D:0.5%],F:3.0%,M:3.7%,n:954 |
|890 Complete BUSCOs (C) |
|885 Complete and single-copy BUSCOs (S) |
|5 Complete and duplicated BUSCOs (D) |
|29 Fragmented BUSCOs (F) |
|35 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Quast also failed again with this error:
Python/2.7.18-GCCcore-10.2.0(58):ERROR:150: Module 'Python/2.7.18-GCCcore-10.2.0' conflicts with the currently loaded module(s) 'Python/3.8.6-GCCcore-10.2.0'
Python/2.7.18-GCCcore-10.2.0(58):ERROR:102: Tcl command execution failed: conflict Python
Python/2.7.18-GCCcore-10.2.0(58):ERROR:150: Module 'Python/2.7.18-GCCcore-10.2.0' conflicts with the currently loaded module(s) 'Python/3.8.6-GCCcore-10.2.0'
Python/2.7.18-GCCcore-10.2.0(58):ERROR:102: Tcl command execution failed: conflict Python
I’m going to add module purge and module load Python/2.7.18-GCCcore-10.2.0 prior to loading quast. I’m also going to comment out lines that have already run successfully. Submitted batch job 310034. Ran in ~45 mins. Below are the results for the hap2 assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:94.0%[S:92.7%,D:1.3%],F:2.9%,M:3.1%,n:954 |
|896 Complete BUSCOs (C) |
|884 Complete and single-copy BUSCOs (S) |
|12 Complete and duplicated BUSCOs (D) |
|28 Fragmented BUSCOs (F) |
|30 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Once again, quast failed to run and I got this error:
ERROR! File not found (contigs): apul.hifiasm.intial.bp.p_ctg.fa
In case you have troubles running QUAST, you can write to quast.support@cab.spbu.ru
or report an issue on our GitHub repository https://github.com/ablab/quast/issues
Please provide us with quast.log file from the output directory.
I’m going to write quast its own script. Quast can be run with or without a reference genome. I’m going to try both, using Amillepora as the reference genome. In the scripts folder: nano initial_quast.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
module purge
module load Python/2.7.18-GCCcore-10.2.0
module load QUAST/5.0.2-foss-2020b-Python-2.7.18
# previously used QUAST/5.2.0-foss-2021b but it failed and produced module conflict errors
echo "Begin quast of primary and haplotypes (initial run) w/ reference" $(date)
quast -t 10 --eukaryote \
apul.hifiasm.intial.bp.p_ctg.fa \
apul.hifiasm.intial.bp.hap1.p_ctg.fa \
apul.hifiasm.intial.bp.hap2.p_ctg.fa \
/data/putnamlab/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta \
-o /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast
echo "Quast complete (initial run); all QC complete!" $(date)
Submitted batch job 310038. I need to read more about quast command line options, as there seem to be a lot of options. Also need to look into including a reference vs not including a reference. Ran super fast but success! Quast is super informative!!!!! The most useful information is in the report.* files. This is from the report.txt file:
All statistics are based on contigs of size >= 500 bp, unless otherwise noted (e.g., "# contigs (>= 0 bp)" and "Total length (>= 0 bp)" include all contigs).
Assembly apul.hifiasm.intial.bp.p_ctg apul.hifiasm.intial.bp.hap1.p_ctg apul.hifiasm.intial.bp.hap2.p_ctg Amil.v2.01.chrs
# contigs (>= 0 bp) 188 275 162 854
# contigs (>= 1000 bp) 188 275 162 851
# contigs (>= 5000 bp) 188 273 162 748
# contigs (>= 10000 bp) 186 271 162 672
# contigs (>= 25000 bp) 166 246 153 545
# contigs (>= 50000 bp) 98 163 124 445
Total length (>= 0 bp) 518528298 481372407 480341213 475381253
Total length (>= 1000 bp) 518528298 481372407 480341213 475378544
Total length (>= 5000 bp) 518528298 481363561 480341213 475052084
Total length (>= 10000 bp) 518514885 481349140 480341213 474498957
Total length (>= 25000 bp) 518188097 480901871 480181619 472383091
Total length (>= 50000 bp) 515726224 477880931 479141568 468867721
# contigs 188 275 162 854
Largest contig 45111900 21532546 22038975 39361238
Total length 518528298 481372407 480341213 475381253
GC (%) 39.05 39.03 39.04 39.06
N50 16268372 12353884 13054353 19840543
N75 13007972 7901416 8791894 1469964
L50 11 15 15 9
L75 20 28 26 23
# N's per 100 kbp 0.00 0.00 0.00 7.79
Look at all of that info! The initial assembly appears to be the best in terms of all the stats. The total length is longer than the Amillepora and the haplotype assemblies. Additionally, it has the largest contig. The Amillepora genome has a higher N50 but the N50 for the primary assembly still looks good. There were 188 contigs generated in the primary assembly. Haplotype 1 assembly had more contigs (275), while haplotype 2 had less (162). Amillepora has 854 contigs which is so high! The primary assembly contig number is much lower than Atenuis (614), Adigitifera (955), or Amillepora (854). Quast, you have converted me <3. My next steps are to play with the -s flag in hifiasm to determine the threshold at which duplicate haplotigs should be purged. The default is 0.55 and Young et al. (2024) ran it with a range of values (0.55, 0.50, 0.45, 0.40, 0.35, 0.30). They found that all worked well to resolve haplotypes, so they stuck with the default of 0.55. They also used the --primary flag, which outputs a primary and alternate assembly as opposed to the primary, hap1 and hap2 assemblies. In their code, they justified this by saying “running hifiasm using the primary flag as we have no real way of knowing if the haplotypes produced are real or not” (line 826).
Starting a run where -s is 0.3 and 0.8. In the /data/putnamlab/jillashey/Apul_Genome/assembly/scripts folder, nano s30_hifiasm.sh:
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/hifiasm
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting assembly with hifiasm" $(date)
hifiasm -o apul.hifiasm.s30 hifi_rr_allcontam_rem.fasta -s 0.3 -t 36 2> apul_hifiasm_allcontam_rem_s30.asm.log
echo "Assembly with hifiasm complete!" $(date)
conda deactivate
Submitted batch job 310048. In the scripts folder, nano s80_hifiasm.sh:
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/hifiasm
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting assembly with hifiasm" $(date)
hifiasm -o apul.hifiasm.s80 hifi_rr_allcontam_rem.fasta -s 0.80 -t 36 2> apul_hifiasm_allcontam_rem_s80.asm.log
echo "Assembly with hifiasm complete!" $(date)
conda deactivate
Submitted batch job 310049
20240329
The s30 script finished running in about 3 days. Now I’m going to run busco and quast for QC on these assemblies. In the scripts folder: nano s30_primary_busco.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s30.bp.p_ctg.gfa > apul.hifiasm.s30.bp.p_ctg.fa
echo "Begin busco on hifiasm-assembled primary fasta with -s 0.30" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s30.bp.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.s30.primary.busco -m genome
echo "busco complete for hifiasm-assembled primary fasta with -s 0.30" $(date)
Submitted batch job 310241. In the scripts folder: nano s30_hap1_busco.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s30.bp.hap1.p_ctg.gfa > apul.hifiasm.s30.bp.hap1.p_ctg.fa
echo "Begin busco on hifiasm-assembled fasta hap1 with -s 0.30" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s30.bp.hap1.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.s30.hap1.busco -m genome
echo "busco complete for hifiasm-assembled fasta hap1 with -s 0.30" $(date)
Submitted batch job 310242. In the scripts folder: nano s30_hap2_busco.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s30.bp.hap2.p_ctg.gfa > apul.hifiasm.s30.bp.hap2.p_ctg.fa
echo "Begin busco on hifiasm-assembled fasta hap2 with -s 0.30" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s30.bp.hap2.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.s30.hap2.busco -m genome
echo "busco complete for hifiasm-assembled fasta hap2 with -s 0.30" $(date)
Submitted batch job 310243. In the scripts folder: nano s30_quast.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
module purge
module load Python/2.7.18-GCCcore-10.2.0
module load QUAST/5.0.2-foss-2020b-Python-2.7.18
# previously used QUAST/5.2.0-foss-2021b but it failed and produced module conflict errors
echo "Begin quast of primary and haplotypes (s30 run) w/ reference" $(date)
quast -t 10 --eukaryote \
apul.hifiasm.s30.bp.p_ctg.fa \
apul.hifiasm.s30.bp.hap1.p_ctg.fa \
apul.hifiasm.s30.bp.hap2.p_ctg.fa \
/data/putnamlab/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta \
-o /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/s30
echo "Quast complete (s30 run); all QC complete!" $(date)
Submitted batch job 310244. So many jobs! The primary busco finished in about an hour. Let’s look at the results.
Busco for primary assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.3%[S:92.9%,D:0.4%],F:3.1%,M:3.6%,n:954 |
|890 Complete BUSCOs (C) |
|886 Complete and single-copy BUSCOs (S) |
|4 Complete and duplicated BUSCOs (D) |
|30 Fragmented BUSCOs (F) |
|34 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Pretty similar to the initial primary assembly, which was the same in completeness (93.3%) but slightly lower in single copy buscos (92% in initial vs 92.9% in s30).
Busco for the hap1 assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.4%[S:92.9%,D:0.5%],F:3.1%,M:3.5%,n:954 |
|891 Complete BUSCOs (C) |
|886 Complete and single-copy BUSCOs (S) |
|5 Complete and duplicated BUSCOs (D) |
|30 Fragmented BUSCOs (F) |
|33 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Almost identical to primary assembly with this flag. Also pretty similar to the initial hap1 assembly.
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.9%[S:93.6%,D:0.3%],F:2.8%,M:3.3%,n:954 |
|896 Complete BUSCOs (C) |
|893 Complete and single-copy BUSCOs (S) |
|3 Complete and duplicated BUSCOs (D) |
|27 Fragmented BUSCOs (F) |
|31 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Once again, pretty similar to the primary and hap1 assemblies, as well as the initial hap2 assembly. So what do these results mean? That the assembly results aren’t really affected by the -s flag? I will have to see what the s80 results look like.
20240401
The s80 script finished a couple of days ago. Now time to assess completeness and what not. In the scripts folder: nano s80_primary_busco.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s80.bp.p_ctg.gfa > apul.hifiasm.s80.bp.p_ctg.fa
echo "Begin busco on hifiasm-assembled primary fasta with -s 0.80" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s80.bp.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.s80.primary.busco -m genome
echo "busco complete for hifiasm-assembled primary fasta with -s 0.80" $(date)
Submitted batch job 310321. In the scripts folder: nano s80_hap1_busco.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s80.bp.hap1.p_ctg.gfa > apul.hifiasm.s80.bp.hap1.p_ctg.fa
echo "Begin busco on hifiasm-assembled fasta hap1 with -s 0.80" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s80.bp.hap1.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.s80.hap1.busco -m genome
echo "busco complete for hifiasm-assembled fasta hap1 with -s 0.80" $(date)
Submitted batch job 310322. In the scripts folder: nano s80_hap2_busco.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data/
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s80.bp.hap2.p_ctg.gfa > apul.hifiasm.s80.bp.hap2.p_ctg.fa
echo "Begin busco on hifiasm-assembled fasta hap2 with -s 0.80" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s80.bp.hap2.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.s80.hap2.busco -m genome
echo "busco complete for hifiasm-assembled fasta hap2 with -s 0.80" $(date)
Submitted batch job 310323
Talked with Hollie last week about possibly assembly the mitochondrial genome for Apul. There are some Apul mito sequences on NCBI, which I’m going to pull and blast against the pacbio raw reads. If there are any hits, I will know that mito sequences are present in the data and its possible to do a mito assembly. There are 16 putative mito sequences for Apul in the NCBI link above. I’m going to pull those sequences and blast them. Make mito folders:
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
mkdir mito
cd mito
Using the putative Apul mito sequences from NCBI, make a fasta file with the 16 sequences. File is called mito_seqs_ncbi.fasta. Similar to what I did with the viral, euk and prok sequences, blast the mito sequences against the raw reads. In the scripts folder: nano blastn_mito_seqs_ncbi.sh
#!/bin/bash
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Build mito seq db" $(date)
makeblastdb -in /data/putnamlab/jillashey/Apul_Genome/assembly/data/mito/mito_seqs_ncbi.fasta -dbtype nucl -out /data/putnamlab/jillashey/Apul_Genome/assembly/data/mito/mito_seqs_ncbi_db
echo "Blasting hifi reads against viral genomes to look for contaminants" $(date)
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -db /data/putnamlab/jillashey/Apul_Genome/assembly/data/mito/mito_seqs_ncbi_db -outfmt 6 -evalue 1e-4 -perc_identity 90 -out mito_hits_rr.txt
echo "Blast complete!" $(date)
Submitted batch job 310324. Checking back a couple hours later. Let’s look at the mito results first:
wc -l mito_hits_rr.txt
12449 mito_hits_rr.txt
head mito_hits_rr.txt
m84100_240128_024355_s2/246617425/ccs NC_081454.1:14774-15130 94.175 103 6 0 6728 6830 308 206 1.49e-39 158
m84100_240128_024355_s2/246617425/ccs NC_081454.1:2434-16341 94.175 103 6 0 6728 6830 12648 12546 1.49e-39 158
m84100_240128_024355_s2/246485798/ccs NC_081454.1:14774-15130 94.175 103 6 0 4066 4168 308 206 8.50e-40 158
m84100_240128_024355_s2/246485798/ccs NC_081454.1:2434-16341 94.175 103 6 0 4066 4168 12648 12546 8.50e-40 158
m84100_240128_024355_s2/248320207/ccs NC_081454.1:14442-14741 92.079 202 14 2 19987 20187 67 267 3.13e-77 283
m84100_240128_024355_s2/248320207/ccs NC_081454.1:2434-16341 92.079 202 14 2 19987 20187 12075 12275 3.13e-77 283
m84100_240128_024355_s2/257166685/ccs NC_081454.1:14442-14741 93.035 201 14 0 16520 16720 67 267 1.18e-80 294
m84100_240128_024355_s2/257166685/ccs NC_081454.1:2434-16341 93.035 201 14 0 16520 16720 12075 12275 1.18e-80 294
m84100_240128_024355_s2/255267441/ccs NC_081454.1:14442-14741 92.537 201 14 1 14988 15187 67 267 1.86e-78 287
m84100_240128_024355_s2/255267441/ccs NC_081454.1:2434-16341 92.537 201 14 1 14988 15187 12075 12275 1.86e-78 287
I need to talk more with Hollie about the mito asssembly because I am still a little confused about this portion.
The busco information for the s80 assembly run also finished. Let’s look at the results!
Primary assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.5%[S:89.8%,D:3.7%],F:3.2%,M:3.3%,n:954 |
|892 Complete BUSCOs (C) |
|857 Complete and single-copy BUSCOs (S) |
|35 Complete and duplicated BUSCOs (D) |
|31 Fragmented BUSCOs (F) |
|31 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Hap1 assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.6%[S:91.4%,D:2.2%],F:3.1%,M:3.3%,n:954 |
|893 Complete BUSCOs (C) |
|872 Complete and single-copy BUSCOs (S) |
|21 Complete and duplicated BUSCOs (D) |
|30 Fragmented BUSCOs (F) |
|31 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Hap2 assembly:
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:92.3%[S:91.0%,D:1.3%],F:2.9%,M:4.8%,n:954 |
|880 Complete BUSCOs (C) |
|868 Complete and single-copy BUSCOs (S) |
|12 Complete and duplicated BUSCOs (D) |
|28 Fragmented BUSCOs (F) |
|46 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Assemblies look quite similar to one another and to the prior assemblies. 89.8% of single copy buscos for the primary assembly is the lowest of all of the assemblies. I’m now going to run quast with the initial, -s 0.30, -s 0.80, and the Amillepora assemblies to compare. In the scripts folder: nano test_quast.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
module purge
module load Python/2.7.18-GCCcore-10.2.0
module load QUAST/5.0.2-foss-2020b-Python-2.7.18
# previously used QUAST/5.2.0-foss-2021b but it failed and produced module conflict errors
echo "Begin quast of initial, s30, and s80 assemblies w/ reference" $(date)
quast -t 15 --eukaryote \
apul.hifiasm.intial.bp.p_ctg.fa \
apul.hifiasm.intial.bp.hap1.p_ctg.fa \
apul.hifiasm.intial.bp.hap2.p_ctg.fa \
apul.hifiasm.s30.bp.p_ctg.fa \
apul.hifiasm.s30.bp.hap1.p_ctg.fa \
apul.hifiasm.s30.bp.hap2.p_ctg.fa \
apul.hifiasm.s80.bp.p_ctg.fa \
apul.hifiasm.s80.bp.hap1.p_ctg.fa \
apul.hifiasm.s80.bp.hap2.p_ctg.fa \
/data/putnamlab/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta \
-o /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/s80
echo "Quast complete" $(date)
Submitted batch job 310352. Finished in about 5 mins. Here’s quast:
All statistics are based on contigs of size >= 500 bp, unless otherwise noted (e.g., "# contigs (>= 0 bp)" and "Total length (>= 0 bp)" include all contigs).
Assembly apul.hifiasm.intial.bp.p_ctg apul.hifiasm.intial.bp.hap1.p_ctg apul.hifiasm.intial.bp.hap2.p_ctg apul.hifiasm.s30.bp.p_ctg apul.hifiasm.s30.bp.hap1.p_ctg apul.hifiasm.s30.bp.hap2.p_ctg apul.hifiasm.s80.bp.p_ctg apul.hifiasm.s80.bp.hap1.p_ctg apul.hifiasm.s80.bp.hap2.p_ctg Amil.v2.01.chrs
# contigs (>= 0 bp) 188 275 162 180 247 167 206 258 189 854
# contigs (>= 1000 bp) 188 275 162 180 247 167 206 258 189 851
# contigs (>= 5000 bp) 188 273 162 180 247 167 206 256 189 748
# contigs (>= 10000 bp) 186 271 162 178 246 166 204 255 187 672
# contigs (>= 25000 bp) 166 246 153 158 219 161 187 235 178 545
# contigs (>= 50000 bp) 98 163 124 92 142 132 120 155 150 445
Total length (>= 0 bp) 518528298 481372407 480341213 504851641 484060404 461127429 558522339 509131880 465604880 475381253
Total length (>= 1000 bp) 518528298 481372407 480341213 504851641 484060404 461127429 558522339 509131880 465604880 475378544
Total length (>= 5000 bp) 518528298 481363561 480341213 504851641 484060404 461127429 558522339 509123034 465604880 475052084
Total length (>= 10000 bp) 518514885 481349140 480341213 504838228 484053588 461119824 558508926 509116218 465589833 474498957
Total length (>= 25000 bp) 518188097 480901871 480181619 504499732 483598713 461019312 558241588 508764673 465424962 472383091
Total length (>= 50000 bp) 515726224 477880931 479141568 502109496 480738474 460002421 555789785 505834173 464397174 468867721
# contigs 188 275 162 180 247 167 206 258 189 854
Largest contig 45111900 21532546 22038975 30476199 22329680 19744096 22038975 22153531 22038975 39361238
Total length 518528298 481372407 480341213 504851641 484060404 461127429 558522339 509131880 465604880 475381253
GC (%) 39.05 39.03 39.04 39.04 39.03 39.04 39.07 39.05 39.03 39.06
N50 16268372 12353884 13054353 16275225 13330421 14742043 14962207 11978068 12847727 19840543
N75 13007972 7901416 8791894 13021168 9796342 9573480 10779388 8114208 6210685 1469964
L50 11 15 15 13 14 14 16 16 14 9
L75 20 28 26 21 24 24 27 29 26 23
# N's per 100 kbp 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 7.79
So much good info!!! The initial assembly still has the largest contig, but the s80 assembly has the longest total lengths. The Amillepora genome still has the best N50 value but the initial assembly also has a good N50. Overall, the initial assembly is the best out of these assemblies. The initial hap2 assembly has the lowest number of contigs (162). Out of the primary assemblies, the s30 assembly has the lowest number of contigs (180), while the initial assembly had 188 contigs and the s80 assembly had 206 contigs.
20240408
Let’s see how many rows have >1000 bit score
awk '{ if ($NF > 1000) count++ } END { print count }' mito_hits_rr.txt
7056
How many rows have a % match >85%?
awk '$3 > 85 {count++} END {print count}' mito_hits_rr.txt
12449
wc -l mito_hits_rr.txt
12449 mito_hits_rr.txt
There are definitely mito sequences in the raw hifi reads. I’ll be using MitoHiFi to assemble the Apul mito genome. This tool is specific for mitogenome assembly from PacBio HiFi reads. After I assemble it, I will remove it from the hifi raw reads before assembly of the nuclear genome. First, I’ll need to install with conda following the instructions on their github.
cd /data/putnamlab/conda
module load Miniconda3/4.9.2
# Clone repo
git clone https://github.com/marcelauliano/MitoHiFi.git
# Create a conda environment with yml file that is inside MitoHiFi/environment
conda env create -n mitohifi_env -f MitoHiFi/environment/mitohifi_env.yml
To activate and run the now-installed mitohifi:
conda activate mitohifi_env
(mitohifi_env) python MitoHiFi/src/mitohifi.py -h
Now we can run mito hifi! Go back to assembly folder and create a mito db folder. I will need to use mitohifi command findMitoReference.py to pull mito references from closely related genomes. Young et al. 2024 pulled 4 mito genomes from NCBI (Platygyra carnosa, Favites abdita, Dipsastraea favus, and the old Orbicella faveolata). He then ran mitohifi for all of them with the Ofav hifi reads, which I’m not really sure why he did that. Maybe because he wanted to create a phylogenetic tree downstream? I’m going to pull the Acropora millepora mito sequences as a reference.
When I try to activate the conda env, I am getting this:
conda activate /data/putnamlab/conda/mitohifi_env
Not a conda environment: /data/putnamlab/conda/mitohifi_env
conda activate /data/putnamlab/conda/MitoHiFi
Not a conda environment: /data/putnamlab/conda/MitoHiFi
Very strange…maybe I just need to load miniconda and run python /data/putnamlab/conda/MitoHiFi/src/findMitoReference.py?
Go to the assembly sequence folder: nano find_mito_ref.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=125GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module purge
module load Miniconda3/4.9.2
echo "Grabbing mito refs from NCBI" $(date)
python /data/putnamlab/conda/MitoHiFi/src/findMitoReference.py --species "Acropora millepora" --email jillashey@uri.edu --outfolder /data/putnamlab/jillashey/Apul_Genome/dbs
echo "Mito grab complete!" $(date)
Submitted batch job 310649. Immediately got this error:
Traceback (most recent call last):
File "/data/putnamlab/conda/MitoHiFi/src/findMitoReference.py", line 23, in <module>
from Bio import Entrez
ModuleNotFoundError: No module named 'Bio'
So I think I do need to activate the environment. Try to create a new env.
cd /data/putnamlab/conda/
conda create -n mitohifi_env -f MitoHiFi/environment/mitohifi_env.yml
WARNING: A conda environment already exists at '/home/jillashey/.conda/envs/mitohifi_env'
Remove existing environment (y/[n])? n
Ooooo I have a super secret conda env. Let’s try to activate it in the script.
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=125GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module purge
module load Miniconda3/4.9.2
conda activate /home/jillashey/.conda/envs/mitohifi_env
echo "Grabbing mito refs from NCBI" $(date)
python findMitoReference.py --species "Acropora millepora" --email jillashey@uri.edu --outfolder /data/putnamlab/jillashey/Apul_Genome/dbs
echo "Mito grab complete!" $(date)
conda deactivate
Immediately got this error:
CommandNotFoundError: Your shell has not been properly configured to use 'conda activate'.
To initialize your shell, run
$ conda init <SHELL_NAME>
Currently supported shells are:
- bash
- fish
- tcsh
- xonsh
- zsh
- powershell
See 'conda init --help' for more information and options.
I need to do conda init but where?
cd /home/jillashey/.conda/envs/mitohifi_env
conda init
no change /opt/software/Miniconda3/4.9.2/condabin/conda
no change /opt/software/Miniconda3/4.9.2/bin/conda
no change /opt/software/Miniconda3/4.9.2/bin/conda-env
no change /opt/software/Miniconda3/4.9.2/bin/activate
no change /opt/software/Miniconda3/4.9.2/bin/deactivate
no change /opt/software/Miniconda3/4.9.2/etc/profile.d/conda.sh
no change /opt/software/Miniconda3/4.9.2/etc/fish/conf.d/conda.fish
no change /opt/software/Miniconda3/4.9.2/shell/condabin/Conda.psm1
no change /opt/software/Miniconda3/4.9.2/shell/condabin/conda-hook.ps1
no change /opt/software/Miniconda3/4.9.2/lib/python3.8/site-packages/xontrib/conda.xsh
no change /opt/software/Miniconda3/4.9.2/etc/profile.d/conda.csh
no change /home/jillashey/.bashrc
No action taken.
Need to figure this out! Here’s what the conda installation portion of their github says:
- Install MitoFinder and/or MITOS outside of Conda.
- Ensure MitoFinder and/or MITOS are added to the PATH before starting the run. Please note that MitoFinder and/or MITOS should be installed separately and made accessible via the PATH environment variable to ensure their proper integration with MitoHiFi. Once those are installed, do:
#Clone MitoHiFi git repo
git clone https://github.com/marcelauliano/MitoHiFi.git
#create a conda environment with our yml file that is inside MitoHiFi/environment
conda env create -n mitohifi_env -f MitoHiFi/environment/mitohifi_env.yml
Add MitoFinder and/or MITOS to the PATH and then activate your mitohifi_env conda environment.
Hmm confused. come back to this.
20240527
It’s been a while. Coming back to installing mitohifi. I emailed Kevin Bryan about it on 4/30 and he said:
“For 2, it looks like you created the conda environment on the login node, instead of in an interactive session, so the compute nodes are not seeing it (remember that the /home directory on the login nodes is separate from the compute nodes for legacy reasons; I hope to fix this eventually).”
So I need to create the conda environment in an interactive session.
cd /data/putnamlab/conda
interactive
Once in the interactive session, clone the github
git clone https://github.com/marcelauliano/MitoHiFi.git
Create a conda environment with the yml file inside MitoHiFi/environment
conda env create -n mitohifi_env -f MitoHiFi/environment/mitohifi_env.yml
Was taking a while to load and then the connection to the server was broken since I was on/off my computer for most of the day doing library prep. Will retry tomorrow when I am on my computer all day
20240610
Back at it. Let’s try to run this as a job so I dont have to sit here all day.
cd /data/putnamlab/conda
mkdir scripts
cd scripts
In the scripts folder: nano load_mitohifi.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=125GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/conda/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
## Attempting to install mitohifi: https://github.com/marcelauliano/MitoHiFi
echo "Start" $(date)
module load Miniconda3/4.9.2
cd /data/putnamlab/conda/
# Go into interactive mode
interactive
# Clone github
git clone https://github.com/marcelauliano/MitoHiFi.git
# Create conda env
conda env create -n mitohifi_env -f MitoHiFi/environment/mitohifi_env.yml
# Activate conda env
conda activate mitohifi_env
# Attempt to run mitohifi
python MitoHiFi/src/mitohifi.py -h
# Deactivate conda env
conda deactivate
echo "End" $(date)
Submitted batch job 320286. Ran for about an hour and completed but I don’t think it installed properly. From the out file:
Preparing transaction: ...working... done
Verifying transaction: ...working... failed
End Mon Jun 10 08:41:25 EDT 2024
From the error file:
CondaVerificationError: The package for pandas located at /home/jillashey/.conda/pkgs/pandas-1.3.5-py37he8f5f7f_0
appears to be corrupted. The path 'lib/python3.7/site-packages/pandas/util/version/__init__.py'
specified in the package manifest cannot be found.
CondaVerificationError: The package for pandas located at /home/jillashey/.conda/pkgs/pandas-1.3.5-py37he8f5f7f_0
appears to be corrupted. The path 'lib/python3.7/site-packages/pandas/util/version/__pycache__/__init__.cpython-37.pyc'
specified in the package manifest cannot be found.
Bleh.
20240611
Loaded env today (conda activate mitohifi_env) and ran python MitoHiFi/src/mitohifi.py -h and somehow it worked????
usage: MitoHiFi [-h] (-r <reads>.fasta | -c <contigs>.fasta) -f
<relatedMito>.fasta -g <relatedMito>.gbk -t <THREADS> [-d]
[-a {animal,plant,fungi}] [-p <PERC>] [-m <BLOOM FILTER>]
[--max-read-len MAX_READ_LEN] [--mitos]
[--circular-size CIRCULAR_SIZE]
[--circular-offset CIRCULAR_OFFSET] [-winSize WINSIZE]
[-covMap COVMAP] [-v] [-o <GENETIC CODE>]
required arguments:
-r <reads>.fasta -r: Pacbio Hifi Reads from your species
-c <contigs>.fasta -c: Assembled fasta contigs/scaffolds to be searched
to find mitogenome
-f <relatedMito>.fasta
-f: Close-related Mitogenome is fasta format
-g <relatedMito>.gbk -k: Close-related species Mitogenome in genebank
format
-t <THREADS> -t: Number of threads for (i) hifiasm and (ii) the
blast search
optional arguments:
-d -d: debug mode to output additional info on log
-a {animal,plant,fungi}
-a: Choose between animal (default) or plant
-p <PERC> -p: Percentage of query in the blast match with close-
related mito
-m <BLOOM FILTER> -m: Number of bits for HiFiasm bloom filter [it maps
to -f in HiFiasm] (default = 0)
--max-read-len MAX_READ_LEN
Maximum lenght of read relative to related mito
(default = 1.0x related mito length)
--mitos Use MITOS2 for annotation (opposed to default
MitoFinder
--circular-size CIRCULAR_SIZE
Size to consider when checking for circularization
--circular-offset CIRCULAR_OFFSET
Offset from start and finish to consider when looking
for circularization
-winSize WINSIZE Size of windows to calculate coverage over the
final_mitogenom
-covMap COVMAP Minimum mapping quality to filter reads when building
final coverage plot
-v, --version show program's version number and exit
-o <GENETIC CODE> -o: Organism genetic code following NCBI table (for
mitogenome annotation): 1. The Standard Code 2. The
Vertebrate MitochondrialCode 3. The Yeast
Mitochondrial Code 4. The Mold,Protozoan, and
Coelenterate Mitochondrial Code and the
Mycoplasma/Spiroplasma Code 5. The Invertebrate
Mitochondrial Code 6. The Ciliate, Dasycladacean and
Hexamita Nuclear Code 9. The Echinoderm and Flatworm
Mitochondrial Code 10. The Euplotid Nuclear Code 11.
The Bacterial, Archaeal and Plant Plastid Code 12. The
Alternative Yeast Nuclear Code 13. The Ascidian
Mitochondrial Code 14. The Alternative Flatworm
Mitochondrial Code 16. Chlorophycean Mitochondrial
Code 21. Trematode Mitochondrial Code 22. Scenedesmus
obliquus Mitochondrial Code 23. Thraustochytrium
Mitochondrial Code 24. Pterobranchia Mitochondrial
Code 25. Candidate Division SR1 and Gracilibacteria
Code
Confused but not going to question it…alright! First, pull mito sequences from NCBI. I’m going to use Acropora millepora and Acropora digitifera.
cd /data/putnamlab/jillashey/Apul_Genome/assembly
mkdir mito
cd mito
mkdir ref_mito_genome/
python /data/putnamlab/conda/MitoHiFi/src/findMitoReference.py --species "Acropora millepora" \
--email jillashey@uri.edu \
--outfolder ref_mito_genome/
python /data/putnamlab/conda/MitoHiFi/src/findMitoReference.py --species "Acropora digitifera" \
--email bdy8@miami.edu \
--outfolder ref_mito_genome/
In the ref_mito_genome folder, Acropora millepora output is NC_081453.1.fasta and NC_081453.1.gb and Acropora digitifera is NC_022830.1.fasta and NC_022830.1.gb. Now we need to constract the mito genome using mitohifi.py. In the /data/putnamlab/jillashey/Apul_Genome/assembly/scripts folder: nano mitohifi_amil.sh
#!/bin/bash
#SBATCH -t 48:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Starting mito assembly with Amillepora refs" $(date)
conda activate mitohifi_env
cd /data/putnamlab/jillashey/Apul_Genome/assembly/mito
python /data/putnamlab/conda/MitoHiFi/src/mitohifi.py -r /data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.fasta \
-f /data/putnamlab/jillashey/Apul_Genome/assembly/mito/ref_mito_genome/NC_081453.1.fasta \
-g /data/putnamlab/jillashey/Apul_Genome/assembly/mito/ref_mito_genome/NC_081453.1.gb \
-t 8 \
-o 5 #invertebrate mitochondrial code
echo "Mito assembly complete!" $(date)
Submitted batch job 320590. Gives me this error:
CommandNotFoundError: Your shell has not been properly configured to use 'conda activate'.
To initialize your shell, run
$ conda init <SHELL_NAME>
Had this issue above. Try running in interactive mode? Job 320591. Okay still failing. Going to try to do the conda init.
echo $SHELL
/bin/bash
conda init bash
conda init bash
no change /opt/software/Miniconda3/4.9.2/condabin/conda
no change /opt/software/Miniconda3/4.9.2/bin/conda
no change /opt/software/Miniconda3/4.9.2/bin/conda-env
no change /opt/software/Miniconda3/4.9.2/bin/activate
no change /opt/software/Miniconda3/4.9.2/bin/deactivate
no change /opt/software/Miniconda3/4.9.2/etc/profile.d/conda.sh
no change /opt/software/Miniconda3/4.9.2/etc/fish/conf.d/conda.fish
no change /opt/software/Miniconda3/4.9.2/shell/condabin/Conda.psm1
no change /opt/software/Miniconda3/4.9.2/shell/condabin/conda-hook.ps1
no change /opt/software/Miniconda3/4.9.2/lib/python3.8/site-packages/xontrib/conda.xsh
no change /opt/software/Miniconda3/4.9.2/etc/profile.d/conda.csh
no change /home/jillashey/.bashrc
No action taken.
Nothing happened. The shell seems to think everything is fine. Let’s check out the bash files.
nano ~/.bashrc
# .bashrc
# Source global definitions
if [ -f /etc/bashrc ]; then
. /etc/bashrc
fi
# Uncomment the following line if you don't like systemctl's auto-paging feature:
# export SYSTEMD_PAGER=
# User specific aliases and functions
# >>> conda initialize >>>
# !! Contents within this block are managed by 'conda init' !!
__conda_setup="$('/opt/software/Miniconda3/4.9.2/bin/conda' 'shell.bash' 'hook' 2> /dev/null)"
if [ $? -eq 0 ]; then
eval "$__conda_setup"
else
if [ -f "/opt/software/Miniconda3/4.9.2/etc/profile.d/conda.sh" ]; then
. "/opt/software/Miniconda3/4.9.2/etc/profile.d/conda.sh"
else
export PATH="/opt/software/Miniconda3/4.9.2/bin:$PATH"
fi
fi
unset __conda_setup
# <<< conda initialize <<<
Everything looks in order. Check conda installation path
ls -l /opt/software/Miniconda3/4.9.2/bin/conda
-rwxr-xr-x. 1 bryank bryank 531 May 13 2021 /opt/software/Miniconda3/4.9.2/bin/conda
echo $PATH
/opt/software/Miniconda3/4.9.2/bin:/opt/software/Miniconda3/4.9.2/condabin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home/jillashey/.local/bin:/home/jillashey/bin
Tried running conda init in /data/putnamlab/conda and /data/putnamlab/conda/MitoHifi but no changes. When I activate the env when I am NOT in an interactive session, it does start to run which is confusing…Need to email Kevin Bryan.
I also need to blast the symbiont genome information. Based on the ITS2 data, the Acropora spp from the Manava site have mostly A1 and D1 symbionts, so I’ll be using the A1 genome and the D1 genome.
20240617
Going to blast to sym genomes now. First download them:
cd /data/putnamlab/jillashey/Apul_Genome/dbs
wget http://smic.reefgenomics.org/download/Smic.genome.scaffold.final.fa.gz
wget https://marinegenomics.oist.jp/symbd/download/102_symbd_genome_scaffold.fa.gz
In the assembly scripts folder: nano blastn_sym.sh
#!/bin/bash
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Build A1 seq db" $(date)
makeblastdb -in /data/putnamlab/jillashey/Apul_Genome/dbs/Smic.genome.scaffold.final.fa -dbtype nucl -out /data/putnamlab/jillashey/Apul_Genome/dbs/A1_db
echo "Build D1 seq db" $(date)
makeblastdb -in /data/putnamlab/jillashey/Apul_Genome/dbs/102_symbd_genome_scaffold.fa -dbtype nucl -out /data/putnamlab/jillashey/Apul_Genome/dbs/D1_db
echo "Blasting hifi reads against symbiont A1 genome to look for contaminants" $(date)
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -db /data/putnamlab/jillashey/Apul_Genome/dbs/A1_db -outfmt 6 -evalue 1e-4 -perc_identity 90 -out sym_A1_contaminant_hits_rr.txt
echo "A1 blast complete! Now blasting hifi reads against symbiont D1 genome to look for contaminants" $(date)
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -db /data/putnamlab/jillashey/Apul_Genome/dbs/D1_db -outfmt 6 -evalue 1e-4 -perc_identity 90 -out sym_D1_contaminant_hits_rr.txt
echo "D1 blast complete!"$(date)
Submitted batch job 323705. Ran in about 1.5 days.
20240619
Cat the sym blast results together and remove anything that has a bit score <1000.
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
cat sym_A1_contaminant_hits_rr.txt sym_D1_contaminant_hits_rr.txt > sym_contaminant_hits_rr.txt
awk '$12 > 1000 {print $0}' sym_contaminant_hits_rr.txt > contaminant_hits_sym_passfilter_rr.txt
wc -l contaminant_hits_sym_passfilter_rr.txt
12 contaminant_hits_sym_passfilter_rr.txt
Pretty clean when bit scores <1000 are removed. Copy this data onto my computer and remove the contaminants in the R script.
Still need to get mito hifi to work.
20240622
TRINITY DID IT!!!!!! She is my hero!!!!! She used a docker singularity install and was able to run it. Here are the output files and folders:
cd /data/putnamlab/tconn/mito
-rw-r--r--. 1 trinity.conn 19K Jun 21 13:58 final_mitogenome.fasta
-rw-r--r--. 1 trinity.conn 33K Jun 21 13:58 final_mitogenome.gb
-rw-r--r--. 1 trinity.conn 275 Jun 21 13:58 contigs_stats.tsv
-rw-r--r--. 1 trinity.conn 1004 Jun 21 13:58 shared_genes.tsv
-rw-r--r--. 1 trinity.conn 48K Jun 21 13:58 final_mitogenome.annotation.png
-rw-r--r--. 1 trinity.conn 48K Jun 21 13:58 contigs_annotations.png
-rw-r--r--. 1 trinity.conn 37K Jun 21 13:58 all_potential_contigs.fa
-rw-r--r--. 1 trinity.conn 20K Jun 21 13:58 coverage_plot.png
-rw-r--r--. 1 trinity.conn 20K Jun 21 13:59 final_mitogenome.coverage.png
drwxr-xr-x. 4 trinity.conn 4.0K Jun 21 13:59 potential_contigs
drwxr-xr-x. 2 trinity.conn 4.0K Jun 21 13:59 contigs_circularization
drwxr-xr-x. 2 trinity.conn 4.0K Jun 21 13:59 final_mitogenome_choice
drwxr-xr-x. 2 trinity.conn 4.0K Jun 21 13:59 reads_mapping_and_assembly
drwxr-xr-x. 2 trinity.conn 4.0K Jun 21 13:59 contigs_filtering
drwxr-xr-x. 2 trinity.conn 4.0K Jun 21 13:59 coverage_mapping
drwxr-xr-x. 2 trinity.conn 4.0K Jun 21 17:55 MitoHifi_out
Let’s look at the output files (explanation of output files is here on the mitohifi github. The final_mitogenome.fasta is the final mitochondria circularized and rotated to start at tRNA-Phe and is 18480 bp long for our genome; the final_mitogenome.gb is the final mitochondria annotated in GenBank format.
head final_mitogenome.fasta
>ptg000003l_rc_rotated
CAAACATTAGGACAATAAGACCTGACTTCATCCAAGTGACAAACCACTGGGTTAAATCTG
TTTTATGTTTAATACACAAATTGACGACGGCCATGCAATACCTGTCAATGAAGGATTCAA
GTTTGGGTAAGGTCTCTCGCGGACTATCGAATTAAACGACACGCTCCTCTAATTAAAACA
GTGAACAGCCAAGTTTTTTGAATTTTAACCTTGCGGTCGTACTACTCAAGCGGAAAATTT
CTGACTTTTTAGGATTGCTTCACATCTTTTTCATTATTTACAGTATAGACTACCAGGGTC
CCTAATCCTGTTTGCTCCCCATACTCTCGTGTTTTAGCCATCACACTATAATCTCAAAAA
TAAATAGTCTTCACGTCTAAAGTTCTTTTTTCTATTTACACATTCCACCGCTACAAAAAA
ATTCCATTTACCTTCTTAAATTATAAAACCCTTTTTAATTAAAACGGCCTATCACACCCT
TTACGCTTTTGCCCACAAAACTAGCCCTTAAGTTTCACCGCGTCTGCTGGCACTTAATTT
The final final_mitogenome.coverage.png shows the sequencing coverage throughout the final mitogenome
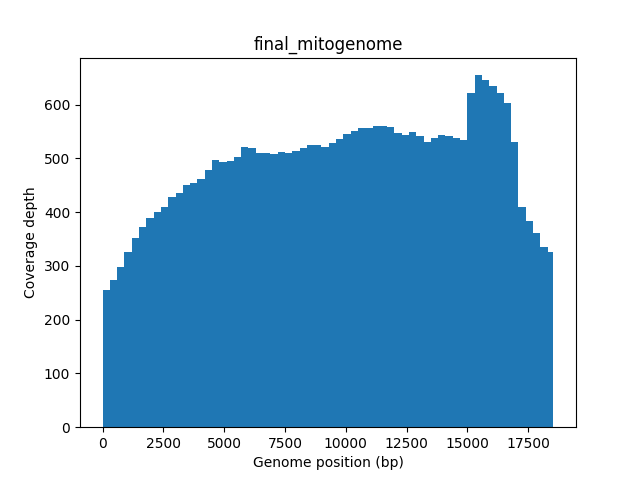
The final final_mitogenome.annotation.png shows the predicted genes throughout the final mitogenome
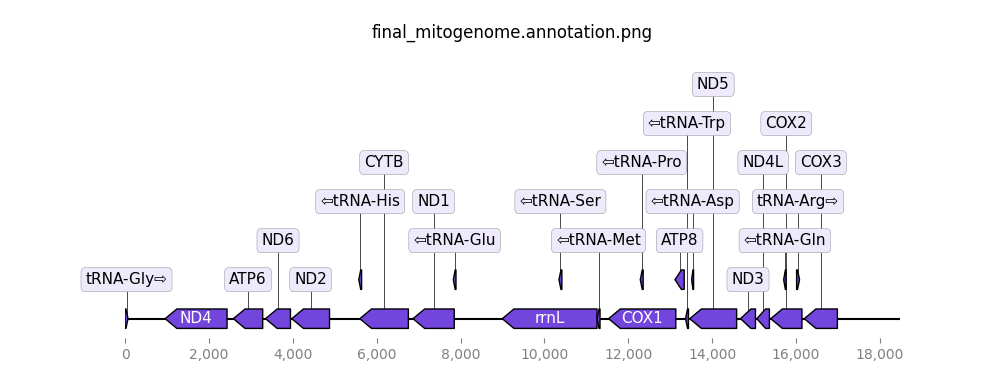
The contigs_stats.tsv file contains the statistics of your assembled mitos such as the number of genes, size, whether it was circularized or not, if the sequence has frameshifts, etc.
less contig_stats.tsv
# Related mitogenome is 18479 bp long and has 17 genes
contig_id frameshifts_found annotation_file length(bp) number_of_genes was_circular
final_mitogenome No frameshift found final_mitogenome.gb 18480 24 True
ptg000003l No frameshift found final_mitogenome.gb 18480 24 True
The shared_genes.tsv shows the comparison of annotation between close-related mitogenome and all potential contigs assembled.
less shared_genes.tsv
contig_id shared_genes unique_to_contig unique_to_relatedMito
final_mitogenome {'ATP6': [1, 1], 'ATP8': [1, 1], 'COX1': [1, 1], 'COX2': [1, 1], 'COX3': [1, 1], 'CYTB': [1, 1], 'ND1': [1, 1], 'ND2': [1, 1], 'ND3': [1, 1], 'ND4': [1, 1], 'ND4L': [1, 1], 'ND5': [1, 1], 'ND6': [1, 1], 'tRNA-Met': [1, 1], 'tRNA-Trp': [1, 1]} {'rrnL': [1, 0], 'tRNA-Arg': [1, 0], 'tRNA-Asp': [1, 0], 'tRNA-Gln': [1, 0], 'tRNA-Glu': [1, 0], 'tRNA-Gly': [1, 0], 'tRNA-His': [1, 0], 'tRNA-Pro': [1, 0], 'tRNA-Ser': [1, 0]} {'l-rRNA': [0, 1], 's-rRNA': [0, 1]}
ptg000003l {'ATP6': [1, 1], 'ATP8': [1, 1], 'COX1': [1, 1], 'COX2': [1, 1], 'COX3': [1, 1], 'CYTB': [1, 1], 'ND1': [1, 1], 'ND2': [1, 1], 'ND3': [1, 1], 'ND4': [1, 1], 'ND4L': [1, 1], 'ND5': [1, 1], 'ND6': [1, 1], 'tRNA-Met': [1, 1], 'tRNA-Trp': [1, 1]} {'rrnL': [1, 0], 'tRNA-Arg': [1, 0], 'tRNA-Asp': [1, 0], 'tRNA-Gln': [1, 0], 'tRNA-Glu': [1, 0], 'tRNA-Gly': [1, 0], 'tRNA-His': [1, 0], 'tRNA-Pro': [1, 0], 'tRNA-Ser': [1, 0]} {'l-rRNA': [0, 1], 's-rRNA': [0, 1]}
I uploaded all of these files onto the Apul genome github in a mito assembly folder. With the completed mito assembly, I can now blast the Apul mito fasta against the hifi reads. In the /data/putnamlab/jillashey/Apul_Genome/assembly/scripts folder: nano blastn_mito.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load BLAST+/2.13.0-gompi-2022a
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Build Apul mito genome db" $(date)
makeblastdb -in /data/putnamlab/tconn/mito/final_mitogenome.fasta -dbtype nucl -out /data/putnamlab/jillashey/Apul_Genome/dbs/mito_db
echo "Blasting hifi reads against mito genome to look for contaminants" $(date)
blastn -query m84100_240128_024355_s2.hifi_reads.bc1029.fasta -db /data/putnamlab/jillashey/Apul_Genome/dbs/mito_db -outfmt 6 -evalue 1e-4 -perc_identity 90 -out mito_contaminant_hits_rr.txt
echo "Mito blast complete!"$(date)
Submitted batch job 324454. Once this is done running, I can purge all the potential contaminants! Ran in about 2.5 hours. Remove all hits <1000.
awk '$12 > 1000 {print $0}' mito_contaminant_hits_rr.txt > contaminant_hits_mito_passfilter_rr.txt
wc -l contaminant_hits_mito_passfilter_rr.txt
1921 contaminant_hits_mito_passfilter_rr.txt
Copy contaminant_hits_mito_passfilter_rr.txt onto computer and identify the reads that are contaminants. This will produce the file all_contam_rem_good_hifi_read_list.txt, which represents the raw hifi reads with the ones marked as contaminants removed. Copy the file all_contam_rem_good_hifi_read_list.txt that was generated from the R script. This specific file was written starting on line 290. It contains the reads that have passed contamination filtering. I copied this file into /data/putnamlab/jillashey/Apul_Genome/assembly/data.
wc -l all_contam_rem_good_hifi_read_list.txt
5896466 all_contam_rem_good_hifi_read_list.txt
Remove the length information from the file
awk '{$2=""; print $0}' all_contam_rem_good_hifi_read_list.txt > output_file.txt
wc -l output_file.txt
5896466 output_file.txt
Run the subseq.sh script in /data/putnamlab/jillashey/Apul_Genome/assembly/scripts to subset the raw hifi fasta file to remove the contaminants identified above. Submitted batch job 324463
20240623
Script above ran in about 22 minutes and the resulting output file is /data/putnamlab/jillashey/Apul_Genome/assembly/data/hifi_rr_allcontam_rem.fasta. This file represents the raw hifi reads with eukaryotic, mitochondrial, symbiont, viral and prokaryotic contaminant reads removed. Out of 5898386 raw hifi reads, there were only 1922 that were identified as contamination. This is only 0.03258519% of the raw reads, which is pretty amazing!
Now that we have clean reads, assembly can begin! In my crazy code above, I ran a couple of different iterations of hifiasm changing the -s option, which sets a similary threshold for duplicate haplotigs that should be purged; the default is 0.55. The iterations that I ran (0.3, 0.55, and 0.8) all worked well to resolve haplotypes with the heterozygosity. Therefore, I stuck with the default 0.55 option. I’m also using -primary to output a primary and alternate assembly, instead of an assembly and two haplotype assemblies, as we have no real way of knowing if the haplotypes produced are real or not.
In the scripts folder, modify hifiasm.sh.
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/hifiasm
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting assembly with hifiasm" $(date)
hifiasm -o apul.hifiasm.s55_pa hifi_rr_allcontam_rem.fasta --primary -s 0.55 -t 36 2> apul_hifiasm_allcontam_rem_s55_pa.log
echo "Assembly with hifiasm complete!" $(date)
conda deactivate
Submitted batch job 324472
20240626
Took about 3 days to run. Yay output!! The primary assembly file is apul.hifiasm.s55_pa.p_ctg.gfa and the alternate assembly file is apul.hifiasm.s55_pa.a_ctg.gfa. Let’s QC! Convert gfa to fa
## PRIMARY
awk '/^S/{print ">"$2"\n"$3}' apul.hifiasm.s55_pa.p_ctg.gfa | fold > apul.hifiasm.s55_pa.p_ctg.fa
zgrep -c ">" apul.hifiasm.s55_pa.p_ctg.fa
187
## ALTERNATE
awk '/^S/{print ">"$2"\n"$3}' apul.hifiasm.s55_pa.a_ctg.gfa | fold > apul.hifiasm.s55_pa.a_ctg.fa
zgrep -c ">" apul.hifiasm.s55_pa.a_ctg.fa
3548
Run busco for the primary and alternate assembly. In the scripts folder: nano busco_qc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
###### PRIMARY ASSEMBLY w/ -s 0.55
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s55_pa.p_ctg.gfa > apul.hifiasm.s55_pa.p_ctg.fa
echo "Begin busco on hifiasm-assembled primary fasta with -s 0.55" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.primary.busco -m genome
echo "busco complete for hifiasm-assembled primary fasta with -s 0.55" $(date)
###### ALTERNATE ASSEMBLY w/ -s 0.55
awk '/^S/{print ">"$2;print $3}' apul.hifiasm.s55_pa.a_ctg.gfa > apul.hifiasm.s55_pa.a_ctg.fa
echo "Begin busco on hifiasm-assembled alternate fasta with -s 0.55" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.a_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.alternate.busco -m genome
echo "busco complete for hifiasm-assembled alternate fasta with -s 0.55" $(date)
Submitted batch job 327004. Got this error:
Use --offline to prevent permission denied issues from downloads
2024-06-27 08:00:19 ERROR: The input file does not contain nucleotide sequences.
2024-06-27 08:00:19 ERROR: BUSCO analysis failed !
2024-06-27 08:00:19 ERROR: Check the logs, read the user guide (https://busco.ezlab.org/busco_userguide.html), and check the BUSCO issue board on https://gitlab.com/ezlab/busco/issues
Convert gfa to fa
## PRIMARY
awk '/^S/{print ">"$2"\n"$3}' apul.hifiasm.s55_pa.p_ctg.gfa | fold > apul.hifiasm.s55_pa.p_ctg.fa
zgrep -c ">" apul.hifiasm.s55_pa.p_ctg.fa
187
## ALTERNATE
awk '/^S/{print ">"$2"\n"$3}' apul.hifiasm.s55_pa.a_ctg.gfa | fold > apul.hifiasm.s55_pa.a_ctg.fa
zgrep -c ">" apul.hifiasm.s55_pa.a_ctg.fa
3548
In the scripts folder: nano quast_qc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
module purge
module load Python/2.7.18-GCCcore-10.2.0
module load QUAST/5.0.2-foss-2020b-Python-2.7.18
# previously used QUAST/5.2.0-foss-2021b but it failed and produced module conflict errors
echo "Begin quast of primary and alternate assemblies w/ reference" $(date)
quast -t 10 --eukaryote \
apul.hifiasm.s55_pa.p_ctg.fa \
apul.hifiasm.s55_pa.a_ctg.fa \
/data/putnamlab/jillashey/genome/Ofav_Young_et_al_2024/Orbicella_faveolata_gen_17.scaffolds.fa \
/data/putnamlab/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta \
/data/putnamlab/jillashey/genome/Aten/GCA_014633955.1_Aten_1.0_genomic.fna \
/data/putnamlab/jillashey/genome/Ahya/GCA_014634145.1_Ahya_1.0_genomic.fna \
/data/putnamlab/jillashey/genome/Ayon/GCA_014634225.1_Ayon_1.0_genomic.fna \
/data/putnamlab/jillashey/genome/Mcap/V3/Montipora_capitata_HIv3.assembly.fasta \
/data/putnamlab/jillashey/genome/Pacuta/V2/Pocillopora_acuta_HIv2.assembly.fasta \
/data/putnamlab/jillashey/genome/Peve/Porites_evermanni_v1.fa \
/data/putnamlab/jillashey/genome/Ofav/GCF_002042975.1_ofav_dov_v1_genomic.fna \
/data/putnamlab/jillashey/genome/Pcomp/Porites_compressa_contigs.fasta \
/data/putnamlab/jillashey/genome/Plutea/plut_final_2.1.fasta \
-o /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast
echo "Quast complete; all QC complete!" $(date)
Run busco for the primary and alternate assembly. In the scripts folder: nano busco_qc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
###### PRIMARY ASSEMBLY w/ -s 0.55
echo "Begin busco on hifiasm-assembled primary fasta with -s 0.55" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.p_ctg.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.primary.busco -m genome
echo "busco complete for hifiasm-assembled primary fasta with -s 0.55" $(date)
###### ALTERNATE ASSEMBLY w/ -s 0.55
#echo "Begin busco on hifiasm-assembled alternate fasta with -s 0.55" $(date)
#labbase=/data/putnamlab
#busco_shared="${labbase}/shared/busco"
#[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.a_ctg.fa" # set this to the query (genome/transcriptome) you are running
#[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
#source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
#cd "${labbase}/${Apul_Genome/assembly/data}"
#busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.alternate.busco -m genome
#echo "busco complete for hifiasm-assembled alternate fasta with -s 0.55" $(date)
Only going to do the primary because was getting erros before in the input file formats. The busco for alternate assembly is commented out. Submitted batch job 327033. Ran successfully in about 30 mins! Here are the results for the primary assembly:
# BUSCO version is: 5.2.2
# The lineage dataset is: metazoa_odb10 (Creation date: 2024-01-08, number of genomes: 65, number of BUSCOs: 954)
# Summarized benchmarking in BUSCO notation for file /data/putnamlab/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.p_ctg.fa
# BUSCO was run in mode: genome
# Gene predictor used: metaeuk
***** Results: *****
C:93.3%[S:92.0%,D:1.3%],F:3.1%,M:3.6%,n:954
890 Complete BUSCOs (C)
878 Complete and single-copy BUSCOs (S)
12 Complete and duplicated BUSCOs (D)
30 Fragmented BUSCOs (F)
34 Missing BUSCOs (M)
954 Total BUSCO groups searched
Dependencies and versions:
hmmsearch: 3.3
metaeuk: GITDIR-NOTFOUND
93.3% completeness, which is the same as my initial/iterative runs. 92% of single copy BUSCOs, which is great for the assembly.
Quast also finished running in about 6 mins. It created a lot of output files:
2024-06-27 09:00:57
RESULTS:
Text versions of total report are saved to /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/report.txt, report.tsv, and report.tex
Text versions of transposed total report are saved to /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/transposed_report.txt, transposed_report.tsv, and transposed_report.tex
HTML version (interactive tables and plots) is saved to /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/report.html
PDF version (tables and plots) is saved to /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/report.pdf
Icarus (contig browser) is saved to /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/icarus.html
Log is saved to /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast/quast.log
Downloaded icarus.html, report.html, report.pdf, and report.txt to /Users/jillashey/Desktop/PutnamLab/Repositories/Apulchra_genome/output/assembly/primary on my personal computer.
20240630
The pipeline from Young et al. 2024 used ragtag and ntlinks to scaffold the assembly. Now I need to do the same. I used hifiasm to assembly the long reads into contigs (approx 168 contigs in the primary assembly). Next, I need to assembly the contigs into scaffolds.
- Contigs = set of partially overlapping reads
- Scffold = set of contigs ordered and oriented to position information. The scaffolds also incorporate empty spaces or gaps.
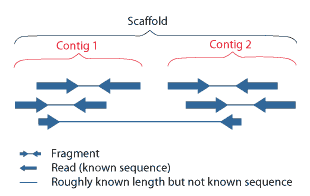
Young et al. 2024 ended up going with the ntlinks to assemble the scaffolds. The ragtag program uses old reference genomes, so Young et al. used the old Orbicella genome assembly and I would have to use one of the old Acropora assemblies. ntlinks uses the long read information along with the newly assembled contigs. I think I will go with ntlink because it is a tool for de novo genome assembly and long read data can be used with it. The ntlink software has options to run multiple iterations/rounds of ntlink to achieve the highest possible contiguity without sacrificing assembly correctness. From the Basic Protocol 3 from the ntlinks paper: “Using the in-code round capability of ntLink allows a user to maximize the contiguity of the final assembly without needing to manually run ntLink multiple times. To avoid re-mapping the reads at each round, ntLink lifts over the mapping coordinates from the input draft assembly to the output post-ntLink scaffolds, which can then be used for the next round of ntLink. The same process can be repeated as many times as needed, thus enabling multiple rounds of ntLink to be powered by a single instance of long-read mapping.” Therefore, I need to turn the scaffolds into contigs. Install ntlinks on Andromeda.
cd /data/putnamlab/conda
module load Miniconda3/4.9.2
conda create --prefix /data/putnamlab/conda/ntlink
conda activate /data/putnamlab/conda/ntlink
conda install -c bioconda -c conda-forge ntlink
In the /data/putnamlab/jillashey/Apul_Genome/assembly/scripts folder: nano ntlinks_5rounds.sh
#!/bin/bash -i
#SBATCH -t 30-00:00:00
#SBATCH --nodes=1 --ntasks-per-node=36
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH --exclusive
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
module load Miniconda3/4.9.2
conda activate /data/putnamlab/conda/ntlink
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Starting scaffolding of hifiasm primary assembly with ntlinks (rounds = 5)" $(date)
ntLink_rounds run_rounds_gaps \
t=36 \
g=100 \
rounds=5 \
gap_fill \
target=apul.hifiasm.s55_pa.p_ctg.fa \
reads=hifi_rr_allcontam_rem.fasta \
out_prefix=apul_ntlink_s55
echo "Scaffolding of hifiasm primary assembly with ntlinks (rounds = 5) complete!" $(date)
Submitted batch job 328341
20240701
ntlink ran in about 4 hours and produced a LOT of output files which I’m not sure what they all mean:
-rw-r--r--. 1 jillashey 11G Jun 30 20:55 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 8.6K Jun 30 20:58 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.trimmed_scafs.agp
-rw-r--r--. 1 jillashey 495M Jun 30 21:14 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
-rw-r--r--. 1 jillashey 8.7K Jun 30 21:14 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 72 Jun 30 21:14 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.scaffolds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 72 Jun 30 21:14 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 76 Jun 30 21:14 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.agp -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 63 Jun 30 21:14 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.verbose_mapping.tsv -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 11G Jun 30 21:33 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 8.3K Jun 30 21:49 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.trimmed_scafs.agp
-rw-r--r--. 1 jillashey 495M Jun 30 22:02 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
-rw-r--r--. 1 jillashey 8.4K Jun 30 22:02 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 106 Jun 30 22:02 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 106 Jun 30 22:02 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 110 Jun 30 22:02 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.agp -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 97 Jun 30 22:02 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.verbose_mapping.tsv -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 11G Jun 30 22:20 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 7.7K Jun 30 22:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.trimmed_scafs.agp
-rw-r--r--. 1 jillashey 495M Jun 30 22:50 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
-rw-r--r--. 1 jillashey 7.7K Jun 30 22:50 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 113 Jun 30 22:50 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 113 Jun 30 22:50 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 117 Jun 30 22:50 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.agp -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 104 Jun 30 22:50 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.verbose_mapping.tsv -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 11G Jun 30 23:08 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 7.7K Jun 30 23:23 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.trimmed_scafs.agp
-rw-r--r--. 1 jillashey 495M Jun 30 23:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
-rw-r--r--. 1 jillashey 7.7K Jun 30 23:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 120 Jun 30 23:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 120 Jun 30 23:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 124 Jun 30 23:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.agp -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 111 Jun 30 23:37 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.verbose_mapping.tsv -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 11G Jun 30 23:55 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
-rw-r--r--. 1 jillashey 7.7K Jul 1 00:11 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.trimmed_scafs.agp
-rw-r--r--. 1 jillashey 495M Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
-rw-r--r--. 1 jillashey 7.7K Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 127 Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 127 Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.ntLink.gap_fill.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa
lrwxrwxrwx. 1 jillashey 131 Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.agp -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.ntLink.scaffolds.gap_fill.fa.agp
lrwxrwxrwx. 1 jillashey 118 Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.verbose_mapping.tsv -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.gap_fill.fa.k32.w100.z1000.verbose_mapping.tsv
lrwxrwxrwx. 1 jillashey 90 Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.5rounds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.ntLink.ntLink.ntLink.ntLink.gap_fill.fa
lrwxrwxrwx. 1 jillashey 70 Jul 1 00:24 apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds.fa -> apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.gap_fill.5rounds.fa
I think the files are representing the iterations of ntlink that was run. For example, files with one ntLink in the file name are from the first iteration, files with two ntLink in the file name are from the second iteration, etc. In the output file, it also gave me a lot of info. It gave me a lot of info on the specific code/parameters for each iteration and then provided me with the final file: Done ntLink rounds! Final scaffolds found in apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds.fa. I now need to run QC on the scaffolded assembly.
In the scripts folder: nano busco_ntlink_qc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Convert from gfa to fasta for downstream use" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
echo "Begin busco on scaffolded assembly" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds.fa" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${Apul_Genome/assembly/data}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apul.ntlink.busco -m genome
echo "busco complete for scaffolded assembly" $(date)
Submitted batch job 328382. Failed, need to rerun
In the scripts folder: nano quast_ntlink_qc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
module purge
module load Python/2.7.18-GCCcore-10.2.0
module load QUAST/5.0.2-foss-2020b-Python-2.7.18
# previously used QUAST/5.2.0-foss-2021b but it failed and produced module conflict errors
echo "Begin quast of scaffolded assemblies w/ references" $(date)
quast -t 10 --eukaryote \
apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds.fa \
apul.hifiasm.s55_pa.p_ctg.fa \
apul.hifiasm.s55_pa.a_ctg.fa \
/data/putnamlab/jillashey/genome/Ofav_Young_et_al_2024/Orbicella_faveolata_gen_17.scaffolds.fa \
/data/putnamlab/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta \
/data/putnamlab/jillashey/genome/Aten/GCA_014633955.1_Aten_1.0_genomic.fna \
/data/putnamlab/jillashey/genome/Ahya/GCA_014634145.1_Ahya_1.0_genomic.fna \
/data/putnamlab/jillashey/genome/Ayon/GCA_014634225.1_Ayon_1.0_genomic.fna \
/data/putnamlab/jillashey/genome/Mcap/V3/Montipora_capitata_HIv3.assembly.fasta \
/data/putnamlab/jillashey/genome/Pacuta/V2/Pocillopora_acuta_HIv2.assembly.fasta \
/data/putnamlab/jillashey/genome/Peve/Porites_evermanni_v1.fa \
/data/putnamlab/jillashey/genome/Ofav/GCF_002042975.1_ofav_dov_v1_genomic.fna \
/data/putnamlab/jillashey/genome/Pcomp/Porites_compressa_contigs.fasta \
/data/putnamlab/jillashey/genome/Plutea/plut_final_2.1.fasta \
-o /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast
echo "Quast complete; all QC complete!" $(date)
Submitted batch job 328389. Move results into new directory:
cd /data/putnamlab/jillashey/Apul_Genome/assembly/output/quast
mkdir ntlink
mv *report* ntlink/
mv basic_stats/ ntlink/
mv icarus* ntlink/
mv quast.log ntlink/
Downloaded icarus.html, report.html, report.pdf, and report.txt to /Users/jillashey/Desktop/PutnamLab/Repositories/Apulchra_genome/output/assembly/ntlink on my personal computer. The quast looks good!!
All statistics are based on contigs of size >= 500 bp, unless otherwise noted (e.g., "# contigs (>= 0 bp)" and "Total length (>= 0 bp)" include all contigs).
Assembly apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds apul.hifiasm.s55_pa.p_ctg apul.hifiasm.s55_pa.a_ctg Orbicella_faveolata_gen_17.scaffolds Amil.v2.01.chrs GCA_014633955.1_Aten_1.0_genomic GCA_014634145.1_Ahya_1.0_genomic GCA_014634225.1_Ayon_1.0_genomic Montipora_capitata_HIv3.assembly Pocillopora_acuta_HIv2.assembly Porites_evermanni_v1 GCF_002042975.1_ofav_dov_v1_genomic Porites_compressa_contigs plut_final_2.1
# contigs (>= 0 bp) 174 187 3548 51 854 1538 2758 1010 1699 474 8186 1933 1071 2975
# contigs (>= 1000 bp) 174 187 3548 51 851 1519 2681 992 1697 474 8186 1933 1071 2933
# contigs (>= 5000 bp) 174 187 3508 51 748 1164 1473 677 1642 461 6821 1933 1071 2138
# contigs (>= 10000 bp) 172 185 3365 51 672 1013 1084 581 1567 447 5755 1142 1071 1951
# contigs (>= 25000 bp) 153 165 2831 48 545 819 769 460 1008 394 4531 831 1054 1650
# contigs (>= 50000 bp) 89 97 1445 40 445 670 581 377 540 203 3258 687 965 1344
Total length (>= 0 bp) 518313916 518458989 482735234 493925641 475381253 403138309 447200179 438047505 780507976 408287534 603805388 485548939 751252456 552020673
Total length (>= 1000 bp) 518313916 518458989 482735234 493925641 475378544 403124220 447138055 438033530 780506390 408287534 603805388 485548939 751252456 551983631
Total length (>= 5000 bp) 518313916 518458989 482572037 493925641 475052084 402086370 443760117 437148684 780327690 408248011 599829750 485548939 751252456 550215806
Total length (>= 10000 bp) 518300503 518445576 481501732 493925641 474498957 400989068 441052114 436458337 779744100 408137131 592561315 478462976 751252456 548844833
Total length (>= 25000 bp) 517996671 518118788 472081622 493863958 472383091 397954288 435913299 434475505 770218970 407100823 571497796 473919972 750872185 543736169
Total length (>= 50000 bp) 515685899 515656915 421575419 493582559 468867721 392444742 429324050 431614917 753567665 400343351 525186424 468943051 747605773 532574147
# contigs 174 187 3548 51 854 1538 2758 1010 1699 474 8186 1933 1071 2975
Largest contig 45111900 45111900 5479021 40246328 39361238 4392697 10924033 11713616 69151359 16633824 1802771 4771691 7905324 3122227
Total length 518313916 518458989 482735234 493925641 475381253 403138309 447200179 438047505 780507976 408287534 603805388 485548939 751252456 552020673
GC (%) 39.05 39.05 39.02 39.49 39.06 38.93 38.97 39.03 39.66 38.11 39.02 38.99 39.13 39.05
N50 17861421 16268372 721379 33295526 19840543 1165953 1584703 3033871 47716837 5167277 171385 1162446 1540036 660708
N75 13936008 13007972 110933 24061036 1469964 537206 753273 1342298 38979999 3166945 85873 575799 817138 325442
L50 10 11 160 7 9 101 86 45 7 24 935 124 140 242
L75 18 20 589 12 23 228 191 98 11 49 2169 272 308 540
# N's per 100 kbp 0.00 0.00 0.00 1.02 7.79 7389.85 7778.63 6736.15 18.07 0.00 6749.89 26684.10 0.00 8717.64
There is an improvement of number of contigs from the initial assembly (187 contigs for initial assembly and 174 contigs for ntlinks cleaned assembly).
20240801
Met w/ Trinity last week and we discussed next steps for genome structural and functional annotation. We decided that she is going to move forward with the funannotate steps. I am going to focus on obtaining methylation data from the PacBio reads because apparently the reads also contain information about the methylation status of the bases. This is a helpful video that explains how pacbio reads have methylation data. Essentially, it uses kinetics to see how far apart the bases are from one another. Hifi sequencing uses a polymerase that incorporates fluorescently labeled nucleotides in real time complementary to a native DNA strand. Epigenetic modifications, like methylation, impact how fast the bases are added. Base modifications can be inferred from per-base pulse width (PW) and inter-pulse duration (IPD) kinetics.

I am now looking into the options for PacBio DNA methylation detection/estimation. I’ve found a few tools so far.
- MethBat - aggregate and analyze CpG methylation calls. There are four main workflows:
- Rare methylation analysis - Identify regions in a single dataset exhibiting a “rare” methylation patterns relative to a collection of background datasets; requires pre-defined regions such as all known CpG islands.
- Cohort methylation analysis - Identify regions exhibiting different methylation patterns between case and control datasets; requires pre-defined regions such as all known CpG islands.
- Segmentation - Segment (or divide) CpGs for an individual dataset into regions with a shared methylation pattern; no pre-defined regions required.
- Signature generation - Identify regions exhibiting different methylation patterns between case and control datasets; no pre-defined regions required.
The first two require pre-defined regions of CpG islands, so I don’t think I can use those. Signature generation appears to require some kind of contrast? like different treatments or something so that might not be the way to go either. Segmentation seems like the best bet at the moment because it doesn’t require any pre-defined regions. However, the input does require the output from pb-CpG-tools, which needs mapped Hifi reads. I do not have mapped Hifi reads because what would I be mapping to??
- Jasmine - predicts 5mC of each CpG site in Pacbio Hifi reads
- This seems like the package to use based on the sequencing data that we have. Input is Pacbio reads with kinetics. I’m not sure if our data (or hifi reads in general) have kinetics automatically. No worries, I will be able to generate Hifi reads with kinetics with
ccs-kinetics-bystrandify, which is an executable in thepbtkpackage. As stated above, base modifications can be inferred from per-base pulse width (PW) and inter-pulse duration (IPD) kinetics soccsuses this information to apply the kinetic information to the reads (this is my understanding). Theccscall requires a bam file but it is easy to turn a fasta into a bam. - Not sure if I should wait until the structural/functional annotation is completed. It would obviously be more meaningful to have the structural and functional annotation information. But I may just run it on my primary and ntlinks assemblies to see how the programs work.
- This seems like the package to use based on the sequencing data that we have. Input is Pacbio reads with kinetics. I’m not sure if our data (or hifi reads in general) have kinetics automatically. No worries, I will be able to generate Hifi reads with kinetics with
So my next steps are:
- Convert my final ntlinks fasta to bam
- Convert my primary assembly fasta to bam
- Run
ccs-kinetics-bystrandifyin thepbtkpackage on both bam files - Install jasmine (see info here)
- Run jasmine!
After looking briefly on the internet, it looks like there aren’t a ton of tools to convert fasta files to bam files. But the original data came as a bam file (m84100_240128_024355_s2.hifi_reads.bc1029.bam). It is totally unfiltered and unassembled. Let’s try to run that? At least run the ccs-kinetics-bystrandify.
Make a new methylation directory
cd /data/putnamlab/jillashey/Apul_Genome
mkdir methylation
cd methylation
mkdir scripts data output
In the scripts folder: nano ccs-kinetics.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=500GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/pbtk
echo "Adding kinetics information to hifi reads" $(date)
ccs-kinetics-bystrandify /data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.bam /data/putnamlab/jillashey/Apul_Genome/methylation/data/apul_hifi_raw_kinetics.bam
echo "Kinetics complete!" $(date)
conda deactivate
Submitted batch job 333762. Pended for an hour, then ran in 5 mins.
20240802
Time to install jasmine!
cd/data/putnamlab/conda
module load Miniconda3/4.9.2
conda create --prefix /data/putnamlab/conda/jasmine
conda install -c bioconda jasmine
This takes a couple of minutes but once its installed, jasmine can be run. In the /data/putnamlab/jillashey/Apul_Genome/methylation/scripts folder, nano jasmine.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/jasmine
echo "Running jasmine" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data/
jasmine apul_hifi_raw_kinetics.bam apul_hifi_raw_kinetics_5mc.bam
echo "Jasmine complete!" $(date)
conda deactivate
Submitted batch job 333785. Failed immediately with this error:
ERROR StatusLogger No log4j2 configuration file found. Using default configuration: logging only errors to the console.
Exception in thread "main" java.lang.NullPointerException
at uio.amg.zhong.jasmine.JASMINE.findXMLfile(JASMINE.java:494)
at uio.amg.zhong.jasmine.JASMINE.main(JASMINE.java:40)
20240829
Hollie and I met earlier this week and we briefly discussed methylation for Apul. I said that some of the MethBat tools required information about known CpG locations. She recommended I try emboss fuzznuc, which searches for specific patterns in nucleotide sequences (such as CGs). Why do we care about CGs? CpG sites occur when a cytosine is followed by a guanine in a linear sequence. Cytosines in CpG motifs can be methylated. So in order to find methylation, we need to find the CpG sites.
The Roberts lab has used the fuzznuc program before to identidy CG motifs; Sam’s notebook has an example of how to use fuzznuc, and this page has instances where the output file from fuzznuc was used in analysis. Emboss is already installed on Andromeda yay. In the /data/putnamlab/jillashey/Apul_Genome/methylation/scripts folder: nano fuzznuc.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
# Load module
module load EMBOSS/6.6.0-foss-2018b
echo "Running fuzznuc on assembled Apul genome" $(date)
# Run fuzznuc
fuzznuc \
-sequence /data/putnamlab/jillashey/Apul_Genome/assembly/data/apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds.fa \
-pattern CG \
-outfile /data/putnamlab/jillashey/Apul_Genome/methylation/output/CGmotif_fuzznuc_Apul.gff \
-rformat gff
echo "Fuzznuc complete" $(date)
Submitted batch job 336307. Ran very fast. Let’s look at the output.
wc -l CGmotif_fuzznuc_Apul.gff
16011621 CGmotif_fuzznuc_Apul.gff
head CGmotif_fuzznuc_Apul.gff
##gff-version 3
##sequence-region ntLink_7 1 182921
#!Date 2024-08-29
#!Type DNA
#!Source-version EMBOSS 6.6.0.0
ntLink_7 fuzznuc nucleotide_motif 47 48 2 + . ID=ntLink_7.1;note=*pat pattern:CG
ntLink_7 fuzznuc nucleotide_motif 50 51 2 + . ID=ntLink_7.2;note=*pat pattern:CG
ntLink_7 fuzznuc nucleotide_motif 97 98 2 + . ID=ntLink_7.3;note=*pat pattern:CG
ntLink_7 fuzznuc nucleotide_motif 99 100 2 + . ID=ntLink_7.4;note=*pat pattern:CG
ntLink_7 fuzznuc nucleotide_motif 124 125 2 + . ID=ntLink_7.5;note=*pat pattern:CG
tail CGmotif_fuzznuc_Apul.gff
ptg000187l fuzznuc nucleotide_motif 16594 16595 2 + . ID=ptg000187l.326;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 16672 16673 2 + . ID=ptg000187l.327;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 16891 16892 2 + . ID=ptg000187l.328;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 16923 16924 2 + . ID=ptg000187l.329;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 17048 17049 2 + . ID=ptg000187l.330;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 17120 17121 2 + . ID=ptg000187l.331;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 17701 17702 2 + . ID=ptg000187l.332;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 17753 17754 2 + . ID=ptg000187l.333;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 17765 17766 2 + . ID=ptg000187l.334;note=*pat pattern:CG
ptg000187l fuzznuc nucleotide_motif 17890 17891 2 + . ID=ptg000187l.335;note=*pat pattern:CG
Lot of instances of CGs in the genome. Calculate how many CG motifs per chromosome.
awk '{print $1}' CGmotif_fuzznuc_Apul.gff | sort | uniq -c > CpG_chrom_counts.txt
174 #!Date
174 ##gff-version
2660 ntLink_0
5528 ntLink_1
18990 ntLink_2
4230 ntLink_3
16135 ntLink_4
718456 ntLink_6
6379 ntLink_7
1131794 ntLink_8
673167 ptg000001l
487373 ptg000002l
481523 ptg000004l
41081 ptg000005l
72434 ptg000006l
384667 ptg000007l
1210027 ptg000008l
576662 ptg000009l
71786 ptg000010l
431224 ptg000011l
612568 ptg000012l
471803 ptg000015l
399662 ptg000016l
402360 ptg000017l
514573 ptg000018l
184959 ptg000019l
546594 ptg000020l
660490 ptg000021l
300528 ptg000022l
1411649 ptg000023l
371263 ptg000024l
651898 ptg000025l
463202 ptg000026l
477238 ptg000027l
20084 ptg000028l
54282 ptg000029c
104517 ptg000030l
487040 ptg000031l
87528 ptg000033l
107979 ptg000034l
294044 ptg000035l
206362 ptg000036l
8351 ptg000037l
9522 ptg000038l
32088 ptg000039l
12627 ptg000040l
3994 ptg000043l
1557 ptg000045l
1134 ptg000046l
379058 ptg000047l
2221 ptg000048l
70943 ptg000049l
822 ptg000050l
11322 ptg000051l
1607 ptg000052l
2012 ptg000053l
976 ptg000054l
1523 ptg000055l
1606 ptg000056l
1168 ptg000057l
55399 ptg000059l
3128 ptg000060c
6757 ptg000061l
962 ptg000063l
4760 ptg000064l
1381 ptg000065l
2038 ptg000066l
5588 ptg000067l
12558 ptg000069l
5742 ptg000070l
31307 ptg000072c
31474 ptg000073l
107 ptg000074l
1752 ptg000075l
10527 ptg000076l
1221 ptg000077l
701 ptg000078l
866 ptg000079l
1273 ptg000080l
5641 ptg000081l
2028 ptg000082l
3914 ptg000083l
3283 ptg000085l
2711 ptg000086l
2651 ptg000087l
2899 ptg000088l
1270 ptg000089l
1474 ptg000090l
797 ptg000092l
1080 ptg000093l
1040 ptg000094l
1193 ptg000095l
1674 ptg000096l
1586 ptg000097l
1601 ptg000098l
1385 ptg000099l
1524 ptg000100l
866 ptg000101l
1850 ptg000102l
2880 ptg000105l
1386 ptg000106l
2113 ptg000107l
2548 ptg000108l
1351 ptg000109l
1268 ptg000112l
1804 ptg000113l
1450 ptg000114l
1189 ptg000115l
1357 ptg000116l
817 ptg000117l
969 ptg000118l
605 ptg000119l
866 ptg000120l
1330 ptg000121l
1436 ptg000122l
1308 ptg000123l
1708 ptg000124l
637 ptg000125l
947 ptg000126l
1023 ptg000127l
1626 ptg000128l
1220 ptg000129l
1187 ptg000130l
812 ptg000131l
855 ptg000132l
1153 ptg000133l
3997 ptg000134l
112 ptg000135l
477 ptg000136l
1906 ptg000137l
2694 ptg000138l
432 ptg000139l
802 ptg000140l
2269 ptg000141l
423 ptg000142l
609 ptg000144l
504 ptg000145l
1177 ptg000146l
1059 ptg000147l
1177 ptg000148l
2720 ptg000149l
4343 ptg000151l
716 ptg000152l
877 ptg000153l
1155 ptg000154l
948 ptg000155l
462 ptg000158l
494 ptg000159l
1126 ptg000160l
408 ptg000161l
311 ptg000162l
388 ptg000163l
151 ptg000164l
782 ptg000165l
360 ptg000166l
519 ptg000167l
392 ptg000168l
535 ptg000169l
468 ptg000170l
504 ptg000171l
1376 ptg000172l
697 ptg000173l
568 ptg000174l
525 ptg000175l
1460 ptg000176l
1564 ptg000177l
554 ptg000178l
1129 ptg000179l
220 ptg000180l
1048 ptg000181l
746 ptg000182l
892 ptg000183l
1494 ptg000184l
1252 ptg000185l
471 ptg000186l
335 ptg000187l
174 ##sequence-region
174 #!Source-version
174 #!Type
20240910
Met with Trinity this morning! She has completed the repeat masker/modeling part and is now working on the structural and functional annotation. We discussed submitting the assembled genome to NCBI, which I am going to look into. When I was making the submission on GenBank, one of the submission questions was “do you want to submit motif/modification information” since its PacBio sequencing. I looked at NCBI’s page on this and they mentioned an analysis workflow (RS_Modification_and_Motif_Analysis) that will identify motifs and modifications but I couldn’t find much info about it on the pacbio website. I emailed the pacbio people to see where I should start my methylation analysis. I was also looking at the Apul PacBio summary report and it has some information on methylation as well which I have never noticed. It has plots of CpG methylation in reads but no other information.
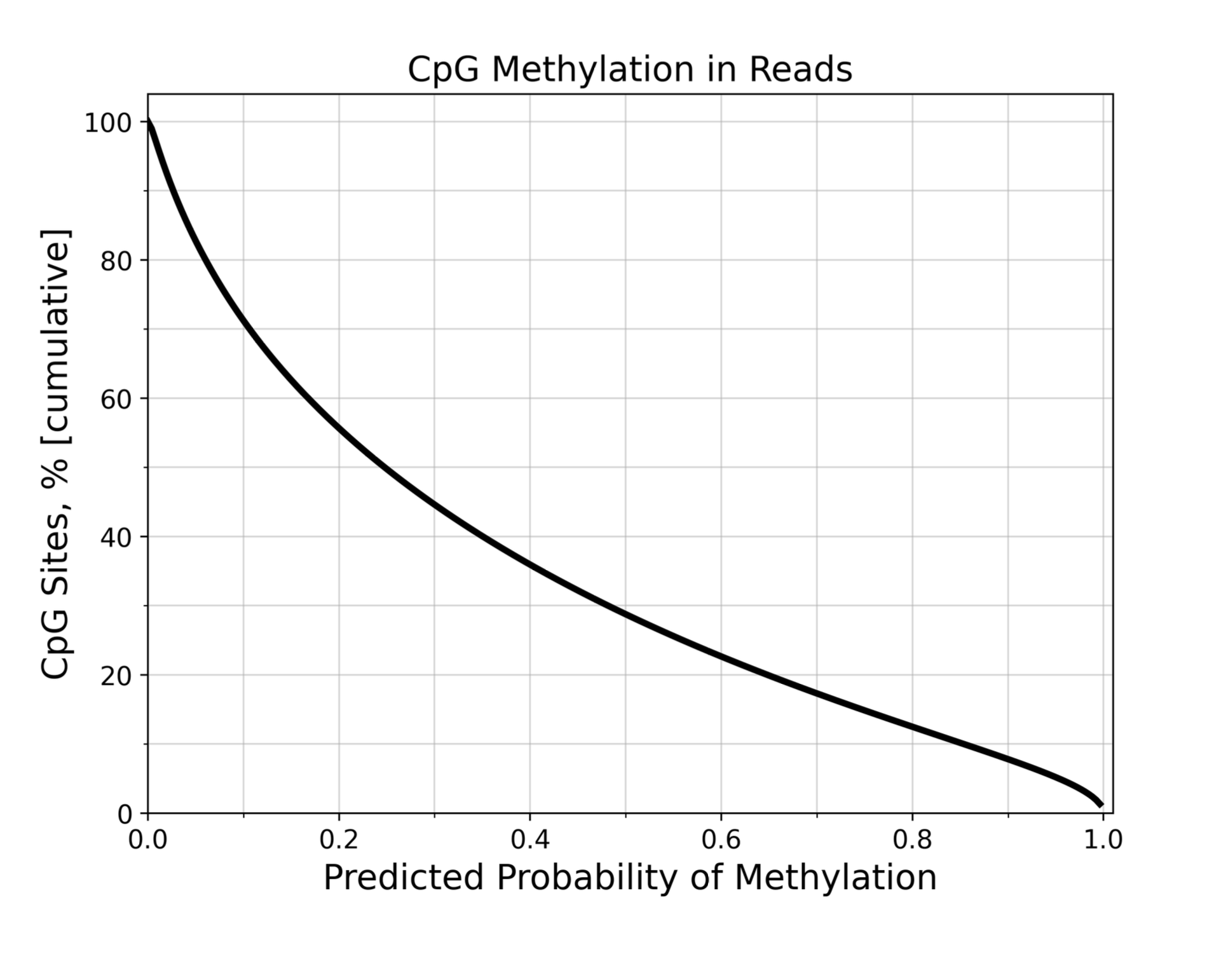
I emailed PacBio to ask where to start with all these things.
20240918
Maybe the raw hifi reads already have the kinetics info in them that is needed for jasmine. Before I tried to convert the original bam file to one with kinetics but now I’m going to try running just the raw hifi bam through jasmine.
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/jasmine
echo "Running jasmine" $(date)
jasmine /data/putnamlab/jillashey/Apul_Genome/assembly/data/m84100_240128_024355_s2.hifi_reads.bc1029.bam /data/putnamlab/jillashey/Apul_Genome/methylation/data/apul_hifi_5mc.bam
echo "Jasmine complete!" $(date)
conda deactivate
Submitted batch job 338453. Failed with same error as before:
bash: cannot set terminal process group (-1): Function not implemented
bash: no job control in this shell
ERROR StatusLogger No log4j2 configuration file found. Using default configuration: logging only errors to the console.
Exception in thread "main" java.lang.NullPointerException
at uio.amg.zhong.jasmine.JASMINE.findXMLfile(JASMINE.java:494)
at uio.amg.zhong.jasmine.JASMINE.main(JASMINE.java:40)
Got email back from PacBio people:
“For your record, we have opened case 00233359 for this inquiry.
First, I think the information on the NCBI page is not going to be as relevant in this particular instance. The base modification files that are referenced on that page are outputs from the Microbial Genome Analysis workflow and they emphasize 6mA and 4mC motifs which are the most common modifications in bacterial genomes.
For methylation analyses in eukaryotes, our key tools are focused on analysis of 5mC in CpG sites. 5mC methylation probabilities in CpG sites are called using our tools primrose or jasmine and are encoded in the hifi_reads.bam file as the MM and ML tags. We have two tools for the analyses of these data pb CpGtools and methbat.
pb cpgtools is the older of the two tools and uses either a trained machine learning model or a pileup model to summarize methylation probabilities across sites to provide evidence of hyper- or hypo-methylation. Pbcpgtools can also be used to summarize 5mC calls for individual samples, which can then be used to build cohort profiles for methbat.
Methbat is the newer of the two tools and is technically still an “in development”, but it has four workflows that are supported, which are summarized here on the user guide page. Which workflow you want to use is going to depend your experimental design and what you would like to test.
If you would be up for sending me some information on your dataset and what you are interested testing for, I would be very happy to weigh in on which workflow might be most useful.”
I sent him some info about the data that I have, what I am trying to do and what code I have tried so far. Going to download the example datasets from the PacBio websites to try to run jasmine in interactive mode. Going to submit as a job.
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/jasmine
echo "Running jasmine" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data/test
jasmine m64168_200820_000733.subreads.bam test_jasmine.bam
echo "Jasmine complete!" $(date)
conda deactivate
Submitted batch job 338456. Hmm got the same error as above…so maybe an installation error?
20240924
PacBio responded with very helpful info about methylation analysis. They recommended that I:
- Align sequences to reference genome with pbmm2, a minimap2 SMRT wrapper specifically for PacBio data.
- Use the aligned bam file as input for pb-CpG-tools which generates site methylation probabilities for hifi reads
- Use pb-CpG-tools output as input for MethBat
I do not need to run ccs-bystrandify or jasmine because the 5mC calling takes place on the instrument, so MM and ML tags should already be included with the data. Let’s install pbmm2 via PacBio conda instructions.
cd /data/putnamlab/conda
module load Miniconda3/4.9.2
conda create --prefix /data/putnamlab/conda/pbmm2
conda activate /data/putnamlab/conda/pbmm2
conda install -c bioconda pbmm2
conda deactivate
The pbmm2 documentation says to align the bam file to the reference genome. But I just created the reference genome from these reads…I guess I will use the new reference genome? I am going to used the unmasked version that is in my folder. In the /data/putnamlab/jillashey/Apul_Genome/assembly/scripts folder: nano pbmm2_index.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/pbmm2
echo "Indexing reference genome" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
pbmm2 index apul.hifiasm.s55_pa.p_ctg.fa.k32.w100.z1000.ntLink.5rounds.fa apul_ref_out.mmi --preset CCS
echo "Index of ref genome complete" $(date)
conda deactivate
Submitted batch job 339851. Took about 15 seconds yay. Now let’s align the raw bam file to the index. I could align either a fasta or bam file to the reference. I’m going to start with the raw bam file. This file does not have any contaminants removed or is assembled in any way. nano pbmm2_align.sh
#!/bin/bash -i
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/assembly/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
conda activate /data/putnamlab/conda/pbmm2
echo "Aligning raw bam" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/assembly/data
pbmm2 align apul_ref_out.mmi m84100_240128_024355_s2.hifi_reads.bc1029.bam out.aligned.bam --sort --preset HIFI
echo "Alignment complete" $(date)
conda deactivate
Submitted batch job 339861. Currently pending because it needs resources. While I wait, I’m going to install the other pacbio packages for methylation analysis. For the pb-CpG-tools, I need to download the release from github and unpack.
cd /data/putnamlab/conda
wget https://github.com/PacificBiosciences/pb-CpG-tools/releases/download/v2.3.2/pb-CpG-tools-v2.3.2-x86_64-unknown-linux-gnu.tar.gz
tar -xzf pb-CpG-tools-v2.3.2-x86_64-unknown-linux-gnu.tar.gz
# Run help option to test binary and see latest usage details:
pb-CpG-tools-v2.3.2-x86_64-unknown-linux-gnu/bin/aligned_bam_to_cpg_scores --help
Great, that was super easy. Install MethBat with conda
cd /data/putnamlab/conda
module load Miniconda3/4.9.2
conda create --prefix /data/putnamlab/conda/methbat
conda activate /data/putnamlab/conda/methbat
conda install -c bioconda methbat
conda deactivate
Success! And alignment is currently running. Ran in about 4 hours. Run pb-CpG-tools to assess methylation site probabilities at CpG sites. In the /data/putnamlab/jillashey/Apul_Genome/methylation/scripts folder: nano pb_cpg_probs.sh
#!/bin/bash
#SBATCH -t 100:00:00
#SBATCH --nodes=1 --ntasks-per-node=10
#SBATCH --export=NONE
#SBATCH --mem=250GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Using aligned bam to generate cpg probabilities" $(date)
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data
/data/putnamlab/conda/pb-CpG-tools-v2.3.2-x86_64-unknown-linux-gnu/bin/aligned_bam_to_cpg_scores \
--bam /data/putnamlab/jillashey/Apul_Genome/assembly/data/out.aligned.bam \
--output-prefix Apul.pbmm2 \
--model /data/putnamlab/conda/pb-CpG-tools-v2.3.2-x86_64-unknown-linux-gnu/models/pileup_calling_model.v1.tflite \
--threads 10
echo "cpg probability prediction complete!" $(date)
Submitted batch job 339998
20240925
The pb-CpG-tools has information on the output. Let’s look at the beginning of the bed file:
head Apul.pbmm2.combined.bed
ntLink_7 3388 3389 15.0 Total 4 0 4 0.0
ntLink_7 3395 3396 8.5 Total 4 0 4 0.0
ntLink_7 3431 3432 3.7 Total 5 0 5 0.0
ntLink_7 3467 3468 4.8 Total 5 0 5 0.0
ntLink_7 3507 3508 4.6 Total 5 0 5 0.0
ntLink_7 3512 3513 5.4 Total 5 0 5 0.0
ntLink_7 3536 3537 3.9 Total 5 0 5 0.0
ntLink_7 3546 3547 4.8 Total 5 0 5 0.0
ntLink_7 3552 3553 7.5 Total 5 0 5 0.0
ntLink_7 3607 3608 4.8 Total 5 0 5 0.0
tail Apul.pbmm2.combined.bed
ptg000187l 16593 16594 73.4 Total 49 36 13 73.5
ptg000187l 16671 16672 93.8 Total 46 44 2 95.7
ptg000187l 16890 16891 92.0 Total 42 39 3 92.9
ptg000187l 16922 16923 89.6 Total 40 36 4 90.0
ptg000187l 17047 17048 76.1 Total 32 25 7 78.1
ptg000187l 17119 17120 95.4 Total 31 30 1 96.8
ptg000187l 17700 17701 94.5 Total 15 15 0 100.0
ptg000187l 17752 17753 89.9 Total 14 13 1 92.9
ptg000187l 17764 17765 91.4 Total 14 13 1 92.9
ptg000187l 17889 17890 79.8 Total 10 8 2 80.0
Columns
- Reference name - contig name from reference genome (I used the genome that I assembled)
- Start coordinate
- End coordinate
- Modification score - modification probability score or the likelihood that the cytosine in the CpG site is methylated. Higher score = higher likelihood of methylation at that site
- Haplotype - total, probabilities combined across all reads
- Coverage - number of reads covering CpG site
- Estimated modified site count - number of CpG sites estimated to be methylated
- Estimated unmodified site count - number of CpG sites estimated to be unmethylated
- Discretized modification probability - ratio of modified to unmodified sites, indicating the confidence that the site is methylated (ie modified) or unmethylated
We are interested in the modification score, which ranges from 0 to 100, with 0 being unmethylated and 100 being fully methylated. Scores that range from 20-90 are considered partially methylated and may be areas of active gene expression or transcriptional plasticity.
THIS IS SO COOL!!!!!!
20240930
I am now interested in the overlap of the CpGs with genomic features. Trinity provided me with the path that I can use for the Apul gff: /data/putnamlab/tconn/predict_results/Acropora_pulchra.gff3. Going to use bedtools intersect to find intersections of genes and CpGs.
cd /data/putnamlab/tconn/predict_results
head Acropora_pulchra.gff3
##gff-version 3
ntLink_0 funannotate gene 1105 7056 . + . ID=FUN_000001;
ntLink_0 funannotate mRNA 1105 7056 . + . ID=FUN_000001-T1;Parent=FUN_000001;product=hypothetical protein;
ntLink_0 funannotate exon 1105 1188 . + . ID=FUN_000001-T1.exon1;Parent=FUN_000001-T1;
ntLink_0 funannotate exon 1861 1941 . + . ID=FUN_000001-T1.exon2;Parent=FUN_000001-T1;
ntLink_0 funannotate exon 2762 2839 . + . ID=FUN_000001-T1.exon3;Parent=FUN_000001-T1;
ntLink_0 funannotate exon 5044 7056 . + . ID=FUN_000001-T1.exon4;Parent=FUN_000001-T1;
ntLink_0 funannotate CDS 1105 1188 . + 0 ID=FUN_000001-T1.cds;Parent=FUN_000001-T1;
ntLink_0 funannotate CDS 1861 1941 . + 0 ID=FUN_000001-T1.cds;Parent=FUN_000001-T1;
ntLink_0 funannotate CDS 2762 2839 . + 0 ID=FUN_000001-T1.cds;Parent=FUN_000001-T1;
Look for intersects in methylation data and gff
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data
interactive
module load BEDTools/2.30.0-GCC-11.3.0
bedtools intersect -a Apul.pbmm2.combined.bed -b /data/putnamlab/tconn/predict_results/Acropora_pulchra.gff3 -wa -wb > Apul_methylation_genome_intersect.bed
head Apul_methylation_genome_intersect.bed
ntLink_7 3388 3389 15.0 Total 4 0 4 0.0 ntLink_7 funannotate gene 79 4679 . + . ID=FUN_002303;
ntLink_7 3388 3389 15.0 Total 4 0 4 0.0 ntLink_7 funannotate mRNA 79 4679 . + . ID=FUN_002303-T1;Parent=FUN_002303;product=hypothetical protein;
ntLink_7 3395 3396 8.5 Total 4 0 4 0.0 ntLink_7 funannotate gene 79 4679 . + . ID=FUN_002303;
ntLink_7 3395 3396 8.5 Total 4 0 4 0.0 ntLink_7 funannotate mRNA 79 4679 . + . ID=FUN_002303-T1;Parent=FUN_002303;product=hypothetical protein;
ntLink_7 3431 3432 3.7 Total 5 0 5 0.0 ntLink_7 funannotate gene 79 4679 . + . ID=FUN_002303;
ntLink_7 3431 3432 3.7 Total 5 0 5 0.0 ntLink_7 funannotate mRNA 79 4679 . + . ID=FUN_002303-T1;Parent=FUN_002303;product=hypothetical protein;
ntLink_7 3467 3468 4.8 Total 5 0 5 0.0 ntLink_7 funannotate gene 79 4679 . + . ID=FUN_002303;
ntLink_7 3467 3468 4.8 Total 5 0 5 0.0 ntLink_7 funannotate mRNA 79 4679 . + . ID=FUN_002303-T1;Parent=FUN_002303;product=hypothetical protein;
ntLink_7 3507 3508 4.6 Total 5 0 5 0.0 ntLink_7 funannotate gene 79 4679 . + . ID=FUN_002303;
ntLink_7 3507 3508 4.6 Total 5 0 5 0.0 ntLink_7 funannotate mRNA 79 4679 . + . ID=FUN_002303-T1;Parent=FUN_002303;product=hypothetical protein;
wc -l Apul_methylation_genome_intersect.bed
15564554 Apul_methylation_genome_intersect.bed
Select genes only
grep -w "gene" Apul_methylation_genome_intersect.bed > Apul_methylation_gene_only_intersect.bed
wc -l Apul_methylation_gene_only_intersect.bed
6246019 Apul_methylation_gene_only_intersect.bed
Count number of genes in genome
cd /data/putnamlab/tconn/predict_results
awk '$3 == "gene"' Acropora_pulchra.gff3 | wc -l
44371
cut -f 3 Acropora_pulchra.gff3 | sort | uniq
CDS
exon
gene
mRNA
tRNA
for reference: https://github.com/hputnam/Meth_Compare?tab=readme-ov-file
20241203
Talked with Ross and Trinity last week to discuss figures and tables to include in the paper. Meeting summary (by Trinity):
- We agreed that I will take over the bulk of responsibility for writing the draft, with Jill & I listed as Co-First Authors
- Our first deadline will be December 6th – I will have a rough draft prepared in the overleaf and will let everyone know when that’s done!
- We will drop figures and such in the shared github – Jill and I will also keep in contact
Figures
- image of pulchra + sampling/geographic distribution (Trinity)
- potential Busco scores (Jill + whatever Trinity can help with!)
- repeat content distribution (Trinity)
- bioanalyzer result/sequence quality statistics (Supplementary?) Tables
- comparison assembly statistics to sanger A.palmata & A.cervicornis, and A.digitifera & A.millepora genomes (Jill)
- description of structural + functional annotation & comparison to other Acropora assemblies (Trinity)
Other tasks for Jill & Trinity
- Jill & Trinity: do more literature searches on use of pacbio for detection of methylation data to provide context for Jill’s methylation analysis
- Jill & Trinity: think a little more about whether we want to include any other non-acroporids in genome comparison
Trinity recently reran busco on our masked genome and it looks beautiful! 96.6% completeness! I now need to run busco on the other genomes to compare completeness. We decided to look at Amillepora, Adigitifera, Acervicornis, and Apalmata (see table).
Amil BUSCO: cd /data/putnamlab/jillashey/genome/Amil_v2.01. In this folder: nano amil_busco.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/genome/Amil_v2.01
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Begin busco on Amil fasta" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/genome/Amil_v2.01/Amil.v2.01.chrs.fasta" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${jillashey/genome/Amil_v2.01}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o amil.busco -m genome
echo "busco complete for Amil" $(date)
Submitted batch job 352146
Adig BUSCO: /data/putnamlab/jillashey/genome/Adig. In this folder: nano adig_busco.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/genome/Adig
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Begin busco on Adig fasta" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/genome/Adig/GCA_014634065.1_Adig_2.0_genomic.fna" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${jillashey/genome/Adig}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o adig.busco -m genome
echo "busco complete for Amil" $(date)
Submitted batch job 352147
I am using recently made Acerv and Apalm genomes. For Acerv:
cd /data/putnamlab/jillashey/genome
mkdir jaAcrCerv1.1
cd jaAcrCerv1.1
wget ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/cax/CAXITW01.fasta.gz
In this folder: nano acerv_busco.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/genome/jaAcrCerv1.1
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Begin busco on Acerv fasta" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/genome/jaAcrCerv1.1/CAXITW01.fasta" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${jillashey/genome/jaAcrCerv1.1}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o acerv.busco -m genome
echo "busco complete for Acerv" $(date)
Submitted batch job 352148. For Apalm:
cd /data/putnamlab/jillashey/genome
mkdir jaAcrPala1.1
cd jaAcrPala1.1
wget ftp://ftp.ebi.ac.uk/pub/databases/ena/wgs/public/cax/CAXIQB01.fasta.gz
In this folder: nano apalm_busco.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/genome/jaAcrPala1.1
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Begin busco on Apalm fasta" $(date)
labbase=/data/putnamlab
busco_shared="${labbase}/shared/busco"
[ -z "$query" ] && query="${labbase}/jillashey/genome/jaAcrPala1.1/CAXIQB01.fasta" # set this to the query (genome/transcriptome) you are running
[ -z "$db_to_compare" ] && db_to_compare="${busco_shared}/downloads/lineages/metazoa_odb10"
source "${busco_shared}/scripts/busco_init.sh" # sets up the modules required for this in the right order
# This will generate output under your $HOME/busco_output
cd "${labbase}/${jillashey/genome/jaAcrPala1.1}"
busco --config "$EBROOTBUSCO/config/config.ini" -f -c 20 --long -i "${query}" -l metazoa_odb10 -o apalm.busco -m genome
echo "busco complete for Apalm" $(date)
Submitted batch job 352149. All ran very fast! Look at results:
# Amil
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:92.7%[S:87.2%,D:5.5%],F:2.6%,M:4.7%,n:954 |
|884 Complete BUSCOs (C) |
|832 Complete and single-copy BUSCOs (S) |
|52 Complete and duplicated BUSCOs (D) |
|25 Fragmented BUSCOs (F) |
|45 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
# Adig
--------------------------------------------------
|Results from dataset metazoa_odb10 |
--------------------------------------------------
|C:93.2%[S:92.8%,D:0.4%],F:3.4%,M:3.4%,n:954 |
|889 Complete BUSCOs (C) |
|885 Complete and single-copy BUSCOs (S) |
|4 Complete and duplicated BUSCOs (D) |
|32 Fragmented BUSCOs (F) |
|33 Missing BUSCOs (M) |
|954 Total BUSCO groups searched |
--------------------------------------------------
Acerv and Apalm failed with this error:
2024-12-03 11:09:13 ERROR: Unable to parse metaeuk results. This typically occurs because sequence headers contain pipes ('|'). Metaeuk uses pipes as delimiters in the results files. The additional pipes interfere with BUSCO's ability to accurately parse the results.To fix this problem remove any pipes from sequence headers and try again.
2024-12-03 11:09:13 ERROR: BUSCO analysis failed!
| Checking sequence headers and yes they do contain | . Edit code for both species so that the “ | ” is changed to a “-“ with the following line of code: |
sed 's/|/-/g' GENOME.fasta > GENOME_modified.fasta
Submitted batch job 352154 for Apalm and Submitted batch job 352155 for Acerv. Zoe also informed me of the Acerv and Apalm genomes that Nick assembled (see his github and paper). I am also going to download those and run busco
NCBI genome accessions are GCA_025960835.2 for A. palmata, GCA_037043185.1 for A. cervicornis version 1, and GCA_041430625.1 for A. cervicornis version 2
20250109
With regards to the methylation data, I want to do the following:
- Percentage of exons, introns, intergentic regions with methylated CpGs
- Patterns of CpG density in all features
interactive
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data
cp /data/putnamlab/jillashey/Apul_Genome/assembly/data/chrom_lengths.txt .
# Remove > at beginning of chromosome name
sed 's/^>//' chrom_lengths.txt > cleaned_chrom_lengths.txt
# Extract genes
awk '$3=="transcript" {
split($0, a, "gene_id ");
split(a[2], b, "\"");
print $1"\t"$4-1"\t"$5"\t"b[2]
}' /data/putnamlab/tconn/annotate_results/Acropora_pulchra.gtf > genes.bed
# Extract exons
awk '$3=="exon" {
split($0, a, "gene_id ");
split(a[2], b, "\"");
print $1"\t"$4-1"\t"$5"\t"b[2]
}' /data/putnamlab/tconn/annotate_results/Acropora_pulchra.gtf > exons.bed
# Extract introns (assuming genes are continuous)
bedtools subtract -a genes.bed -b exons.bed > introns.bed
# Change cleaned_chrom_lengths.txt to tab delimited file instead of space delimited file
sed 's/ /\t/' cleaned_chrom_lengths.txt > tab_delimited_chrom_lengths.txt
# Sort tab delim file
sort -k1,1 tab_delimited_chrom_lengths.txt > sorted_chrom_lengths.txt
# Sort gene.bed file
sort -k1,1 -k2,2n genes.bed > sorted_genes.bed
# Extract intergenic regions
bedtools complement -i sorted_genes.bed -g tab_delimited_chrom_lengths.txt > intergenic.bed
Sort methylation data file
sort -k1,1 -k2,2n Apul.pbmm2.combined.bed > sorted_Apul.pbmm2.combined.bed
Intersect bed feature files with methylation data. I need to run these as a job. In the scripts folder: nano intersect_methylation.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Intersect bed feature files with methylation data" $(date)
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data
# Sort methylation file
sort -k1,1 -k2,2n Apul.pbmm2.combined.bed > sorted_Apul.pbmm2.combined.bed
# Intersect
bedtools intersect -a genes.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > gene_methylation.bed
bedtools intersect -a exons.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > exon_methylation.bed
bedtools intersect -a introns.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > intron_methylation.bed
bedtools intersect -a intergenic.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > intergenic_methylation.bed
echo "Intersection complete" $(date)
Submitted batch job 354769. Ran in <5 mins. Count CpGs in each region
wc -l gene_methylation.bed exon_methylation.bed intron_methylation.bed intergenic_methylation.bed
20250124
We are also interested in finding the % of CpGs found in each region, a la Trigg et al. 2021. I need to identify the upstream and downstream flanks of the genes (1000bp on each flank). This also means I need to redefine the intergenic regions.
interactive
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data
rm intergenic*
# Extract 1000bp upstream flanking regions
bedtools flank -i sorted_genes.bed -g tab_delimited_chrom_lengths.txt -l 1000 -r 0 > upstream_flank.bed
# Extract 1000bp downstream flanking regions
bedtools flank -i sorted_genes.bed -g tab_delimited_chrom_lengths.txt -l 0 -r 1000 > downstream_flank.bed
# Combine genes and flanking regions
cat sorted_genes.bed upstream_flank.bed downstream_flank.bed | sort -k1,1 -k2,2n | bedtools merge -i - > genes_with_flanks.bed
# Redefine intergenic regions
bedtools complement -i genes_with_flanks.bed -g sorted_chrom_lengths.txt > updated_intergenic.bed
Intersect methylation calls with upstream flanks, downstream flanks, and updated intergenic regions. In the scripts folder: nano update_intersect_methylation.sh
#!/bin/bash
#SBATCH -t 24:00:00
#SBATCH --nodes=1 --ntasks-per-node=15
#SBATCH --export=NONE
#SBATCH --mem=100GB
#SBATCH --mail-type=BEGIN,END,FAIL #email you when job starts, stops and/or fails
#SBATCH --mail-user=jillashey@uri.edu #your email to send notifications
#SBATCH --account=putnamlab
#SBATCH -D /data/putnamlab/jillashey/Apul_Genome/methylation/scripts
#SBATCH -o slurm-%j.out
#SBATCH -e slurm-%j.error
echo "Intersect flanks and intergenic feature files with methylation data" $(date)
module load BEDTools/2.30.0-GCC-11.3.0
cd /data/putnamlab/jillashey/Apul_Genome/methylation/data
# Sort methylation file
#sort -k1,1 -k2,2n Apul.pbmm2.combined.bed > sorted_Apul.pbmm2.combined.bed
bedtools intersect -a updated_intergenic.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > intergenic_methylation.bed
bedtools intersect -a upstream_flank.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > upstream_flanks_methylation.bed
bedtools intersect -a downstream_flank.bed -b sorted_Apul.pbmm2.combined.bed -wa -wb > downstream_flanks_methylation.bed
echo "Intersection complete" $(date)
Submitted batch job 356222